


Abstract
BACKGROUND: The 2013–2016 Ebola virus outbreak in West Africa was the most widespread in history. In response, alive attenuated recombinant vesicular stomatitis virus (rVSV) vaccine expressing Zaire Ebolavirus glycoprotein (rVSVΔG-ZEBOV-GP) was evaluated in humans.
METHODS: In a phase 1, randomized, dose-ranging, observer-blind, placebo-controlled trial, healthy adults aged 18–65 years were randomized into 4 groups of 10 to receive one of 3 vaccine doses or placebo. Follow-up visits spanned 180 days postvaccination for safety monitoring, immunogenicity testing and any rVSV virus shedding.
RESULTS: Forty participants were injected with rVSVΔG-ZEBOV-GP vaccine (n = 30) or saline placebo (n = 10). No serious adverse events related to the vaccine or participant withdrawals were reported. Solicited adverse events during the 14-day follow-up period were mild to moderate and self-limited, with the exception of injection-site pain and headache. Viremia following vaccination was transient and no longer detectable after study day 3, with no virus shedding in saliva or urine. All vaccinated participants developed serum immunoglobulin G (IgG), as measured by Ebola virus envelope glycoprotein-based enzyme-linked immunosorbent assay (ELISA). Immunogenicity was comparable across all dose groups, and sustained IgG titers were detectable through to the last visit, at study day 180.
INTERPRETATION: In this phase 1 study, there were no safety concerns after a single dose of rVSVΔG-ZEBOV-GP vaccine. IgG ELISA showed persistent high titers at 180 days postimmunization. There was a period of reactogenicity, but in general, the vaccine was well tolerated. This study provides evidence of the safety and immunogenicity of rVSVΔG-ZEBOV-GP vaccine and importance of its further investigation. Trial registration: Clinical-Trials.gov no., NCT02374385
The 2013–2016 Ebola virus disease (EVD) outbreak in West Africa was the most widespread in history.1 The outbreak started in December 2013 in Guinea and rapidly expanded to other countries in the region, with > 11 000 deaths in nearly 30 000 cases.2 At the onset of the outbreak, several Ebola vaccines were under development; however, few had progressed to clinical trials.
An EVD vaccine, rVSVΔG-ZEBOV-GP, was developed at the Canadian National Microbiology Laboratory of the Public Health Agency of Canada using the live, attenuated recombinant vesicular stomatitis virus (rVSV) backbone.3 Consequent to substituting its glycoprotein (GP) gene with that of a target pathogen, live, attenuated rVSV synthesizes and expresses foreign viral GP antigens, which subsequently induce cellular and humoral immunity.4 Wild-type VSV infects primarily cattle and horses, but rarely causes clinical infections in humans.5,6 In vivo, the rVSV is highly attenuated and nonpathogenic, with narrower cell tropism than wild-type VSV;7,8 it replicates normally, expressing the imported GP that binds to host cell receptors and initiates immune responses.9
rVSVΔG-ZEBOV-GP provides both pre- and postchallenge protection in animal models.10,11 In nonhuman primates, the vaccine was well tolerated12 and protective against lethal Zaire Ebola virus (ZEBOV) challenges following a single dose.13 The vaccine induced protective humoral and cellular immune responses in all vaccinated monkeys.14
As part of a coordinated, international effort to expeditiously evaluate candidate EVD vaccines and make them available to control the epidemic, we conducted a phase 1 trial of the rVSVΔG-ZEBOV-GP. The main objective of this coordinated partnership was to determine the lowest vaccine virus dose that would be safe and well tolerated, and induce an immune response.
Methods
Study design and participants
This was a single-centre, randomized, observer-blind, dose-ranging, placebo-controlled trial to assess the safety (including rVSV viremia and shedding) and immune response after a single injection of one of 3 dose levels of the rVSVΔG-ZEBOV-GP vaccine.15
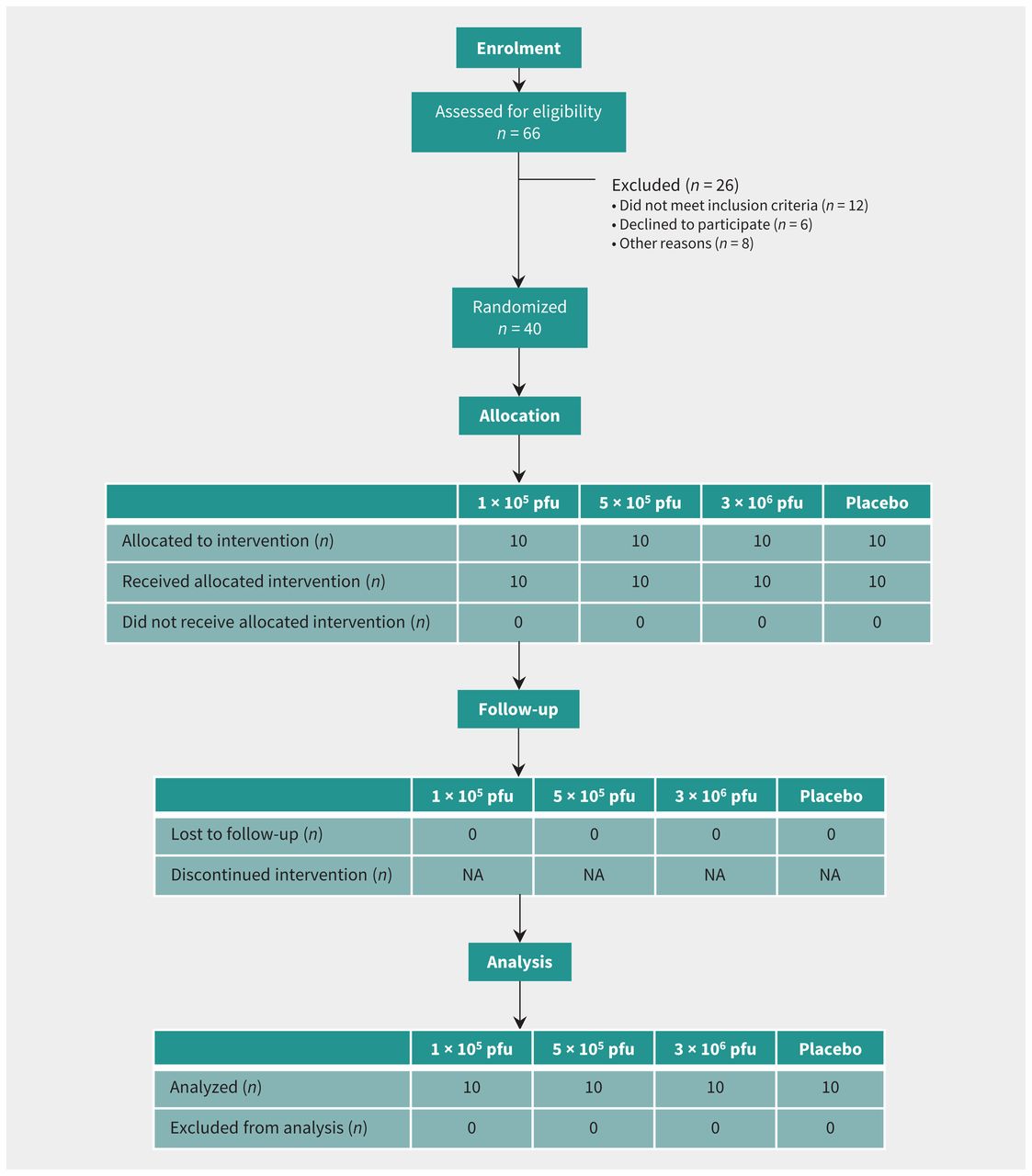
Healthy adult male and female volunteers aged 18 to 65 years were randomly assigned in a 1:1:1:1 ratio to one of 3 vaccine groups (1 × 105 plaque-forming units [pfu], 5 × 105 pfu, and 3 × 106 pfu) or a placebo control group (Figure 1). A randomization list was computer generated with a block size of 8. Exclusions to enrolment included prior infection with a filovirus or VSV or risk of exposure to either; health care worker; child care worker or household contact with young children; pregnant or lactating women; immunocompromised individuals; allergy or any adverse reactions to the vaccine components or other vaccines; underlying medical conditions; and abnormalities on screening tests. Participant screening included medical history, physical examination, electrocardiogram, and blood testing, including complete blood count with white blood cell differential, prothrombin and partial thromboplastin times, serum metabolic panel, urine pregnancy screen (females only), viral serologies (hepatitis B, C, HIV) and urinalysis. The study was conducted at the Canadian Center for Vaccinology in Halifax (clinicaltrials.gov, no. NCT02374385).

Trial diagram showing subjects randomized to vaccine cohorts or placebo. Note: NA = not applicable, pfu = plaque-forming units.
Vaccine
The rVSVΔG-ZEBOV-GP vaccine is a live, attenuated recombinant virus in which the GP gene of VSV Indiana strain is replaced by that of the ZEBOV Kikwit 1995 strain. The vaccine was licensed to BioProtection Systems (NewLink Genetics Corporation) and more recently sublicensed to Merck & Co., Inc. Vaccine product was compliant with good manufacturing practices, suspended in recombinant human serum albumin 2.5 g/L and 10 mM Tris (pH 7.2), dispensed at 1 × 108 pfu/mL per unit vial and stored at −70°C (lot number 0030513). Preservative-free normal saline was used as diluent (Alveda NaCl 0.9% lot number: 13331012) to prepare lower doses. Placebo injections were 1 mL normal saline. Study pharmacists prepared allocated treatment; an unblinded nurse concealed and administered it. The pharmacists did not have any interaction with study participants or blinded study staff, and the unblinded nurse had no other role in the study.
Study procedures
Each participant was injected intramuscularly with 1 mL of vaccine or placebo. Participants were monitored at least 30 minutes postinjection for adverse events (AEs). Assessment visits occurred on days 1, 3, 7, 14, 28, 56, 84 and 180. During the 14-day period after injection, solicited and unsolicited AEs were collected using memory aids; subjects recorded temperature, injection-site reactions, serious adverse events (SAEs) or systemic reactogenicity symptoms. Solicited symptoms included injection-site redness, swelling or pain; subjective and objective fever; chills; sweats; myalgia; arthralgia; fatigue; headache; and gastrointestinal symptoms. Thereafter, unsolicited AEs were documented on days 28, 56, 84 and 180. SAEs were monitored throughout the study. The investigator (SAH) assessed all AEs for causality.
The first volunteer was injected only after day 7 data from the first and lowest dose (3 × 106 pfu) cohort of a separate dose-escalating Walter Reed Army Institute of Research (WRAIR) study16 were evaluated by the independent Data Safety Monitoring Board. This assured us that we could proceed with our dose-ranging design, as 3 × 106 pfu was the highest dose used in this study. Other safety measures included staggered vaccination, follow-up, holding rules and safety monitoring for AEs and SAEs. The study was monitored by an Independent Research Monitor.
Outcomes
The primary outcome was vaccine safety and tolerability through assessment of injection-site events, AEs, SAEs at all visits, and hematologic and biochemical laboratory measures at days 0, 1, 3, 7, 28 and 180, as well as testing for viremia and viral shedding on study days 0–14. Secondary outcomes were antibody measurements on days 0, 7, 14, 28, 56, 84 and 180 by enzyme-linked immunosorbent assay (ELISA), and days 0 and 28 by pseudovirion neutralization assay (PsVNA) 50 and PsVNA80.
An ELISA using recombinant glycoprotein (rGP) from homologous Zaire-Kikwit strain as the solid phase assessed total anti-GP immunoglobulin G (IgG) in sera (Battelle Biomedical Research Center, West Jefferson, OH), Battelle Standard Operating Procedure, BBRC. X-127.17–19 A PsVNA measured neutralizing antibody responses to Ebola GP (US Army Medical Research Institute of Infectious Diseases, Fort Detrick, MD).16,20–24 This assay is based on non-replicating VSV expressing luciferase reporter protein particles pseudotyped with ZEBOV envelope glycoproteins.20 The assay determined the highest serum dilutions causing 50% (PsVNA50) and 80% (PsVNA80) inhibition of virus cell entry and expression of luciferase.
Real-time reverse-transcriptase polymerase chain reaction (RT-PCR) was performed to identify vaccine rVSV in plasma, saliva or urine through amplification of the VSV-nucleoprotein gene.16
Routine hematologic, biochemistry and screening serology were performed at the IWK Health Centre using standard methods.
Statistical analysis
Categorical variables were summarized by number and percentage of participants within each category (with a category for missing data) of the parameter. For continuous variables, the number of participants, mean (or geometric mean, where applicable), median, standard deviation (SD) and 95% confidence interval (CI) for the geometric mean, minimum and maximum values were performed. Statistical hypothesis testing of primary and secondary immunogenicity outcomes was conducted at a 2-sided 0.05 significance level; p values were not adjusted for multiplicity. Summary statistics were performed as well as 2-sided 95% CIs on selected parameters. An analysis of variance (ANOVA) regression model was performed to compare log (base 10) transformed ZEBOV IgG concentrations (in ELISA units/mL); pairwise comparisons of antibody concentrations at analysis days 14, 28 and 180 between rVSVΔG-ZEBOV-GP dose levels and between each rVSVΔG-ZEBOV-GP dose level with placebo were performed in the per-protocol population. The ELISA lower limit of quantification was 55.34 units/mL; samples with a concentration at or below the lower limit of quantification were entered as 27.67 for statistical analysis. PsVNA positivity was defined by titers of 20 or higher; values < 20 were assigned 10 for calculation. For both assays, seroconversion was defined as at least 4-fold increase from baseline titer. Antibody titer calculations were on the log10 scale. Summaries were provided for each vaccine dose and for placebo.
All descriptive statistical analyses were generated using SAS software version 9.3 or later. (SAS and all other SAS Institute Inc. product or service names are registered trademarks or trademarks of SAS Institute Inc., Cary, NC.) Medical history and AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 17.0. Concomitant medications were coded using the most recently available version of the World Health Organization Drug Dictionary.
Ethics approval
Written, informed consent was obtained before any study procedure. The protocol was approved by Health Canada and the Research Ethics Boards of the IWK Health Centre and the Public Health Agency of Canada.
Results
Study participants
A total of 40 participants were vaccinated between Nov. 27 and Dec. 15, 2014 (Table 1). Mean age was 35.6 years (range 18–62) among vaccine recipients, and 32.0 years (range 20–50) among placebo recipients; 23 were female (57.5%). All participants completed follow-up visits (Figure 1).
Baseline characteristics by intervention groups*
Safety
There were no SAEs related to the vaccine or withdrawals from the study. The 3 SAE reports from 2 study subjects were cholelithiasis in a recipient of 5 × 105 pfu vaccine, and psychotic disorder and major depression in a placebo recipient. All were assessed as being unrelated to the vaccine, and were resolved. Reports of AEs solicited from the participants were primarily characterized as mild to moderate, with 3 separate severe events: headache and diarrhea in the 5 × 105 group, and fatigue in the 3 × 106 group. Headache was the most frequent systemic reaction overall (Figure 2). There were no reports of injection-site swelling or redness; only mild to moderate pain was noted locally. In the first day postvaccination, there was higher incidence of injection-site pain in vaccine recipients compared with placebo. Incidence of injection-site pain was 0%, 30% and 80% in the 1 × 105 pfu, 5 × 105 pfu and 3 × 106 pfu cohorts, respectively, and 20% in placebo.

Frequency of local and systemic adverse events (AEs) among vaccine cohorts or placebo. Note: Solicited AEs and their severity reported in the 14 days postinjection for vaccine doses of 1 × 105 plaque-forming units (pfu) (cohort 1), 5 × 105 pfu (cohort 2), 3 × 106 pfu (cohort 3) and placebo shown with 95% confidence intervals for any grade of AEs within each group.
Arthralgia and myalgia had a similar trend, with frequency on the first day postvaccination higher in vaccine than in placebo recipients. Arthralgia was reported in 10%, 0% and 50% in the 1 × 105 pfu, 5 × 105 pfu, and 3 × 106 pfu dose cohorts, respectively, and 0 in placebo; and the incidence of myalgia was 20%, 0% and 40% in the 1 × 105 pfu, 5 × 105 pfu and 3 × 106 pfu cohorts, respectively, and 10% in placebo. Onset of systemic solicited AEs ranged from as early as the day of injection to 14 days thereafter, lasting 1 to 9 days. Objective fever was not measured in any of the participants. A grade 1 subjective fever (3.3%) was reported by 1 participant who received the 3 × 106 pfu dose, and 2 placebo recipients reported grade 1 and grade 2 subjective fever.
Overall, 60% of vaccinees reported unsolicited AEs, yet the incidence of each event was not greater than 2 subjects in any treatment arm (data not shown). Unsolicited AEs were mostly mild to moderate, except a report of severe arthralgia that started at day 18 and lasted 1 day, with no objective signs of arthritis upon immediate examination. No cases of arthritis were observed; however, 1 subject receiving the 3 × 106 pfu dose reported grade 1 joint swelling for 14 days, starting at day 13 postvaccination.
Detection of viremia and virus shedding by PCR
Two participants in the 3 × 106 pfu group developed viremia by the first day postvaccination, which continued to day 3 for 1 of them. Overall, viremia peaked on study day 3, when 18 (60%) of the 30 vaccinated participants were positive, with the greatest frequency in the highest dose group. All subsequent samples were negative (Table 2). There was no virus shedding in saliva or urine.
Polymerase chain reaction detection of rVSVΔG-ZEBOV-GP vaccine virus
Immunogenicity
ZEBOV rGP ELISA
Seroconversions were observed by day 14 in all 3 vaccine dose groups, 4 of 10 (40%) who received the 1 × 105 pfu, 2 of 10 (20%) who received the 5 × 105 pfu, and 4 of 10 (40%) who received the 3 × 106 pfu doses (Table 3). By day 28, seroconversions had increased to 7 of 10 (70%) who received the 1 × 105 pfu or 5 × 105 pfu, and all 10 (100%) who received the 3 × 106 pfu doses. With the exception of 1 subject in the 1 × 105 pfu group, all vaccinees showed seroconversion at some point during the course of the study. Geometric mean titers (GMTs) increased over time from day 0 to day 180, with the exception of a decrease in the 3 × 106 pfu dose group from day 28 to day 180. At day 28, there was a nonsignificant trend to higher GMTs in the 3 × 106 pfu group compared with the low and medium doses (p = 0.071 and p = 0.054, respectively). Serum IgG titers at study day 180 remained significantly higher in all 3 dose groups compared with placebo. GMT responses were similar in the 1 × 105 pfu and 5 × 105 pfu dose groups.
Geometric mean antibody titers to Ebola glycoprotein
PsVNA titers
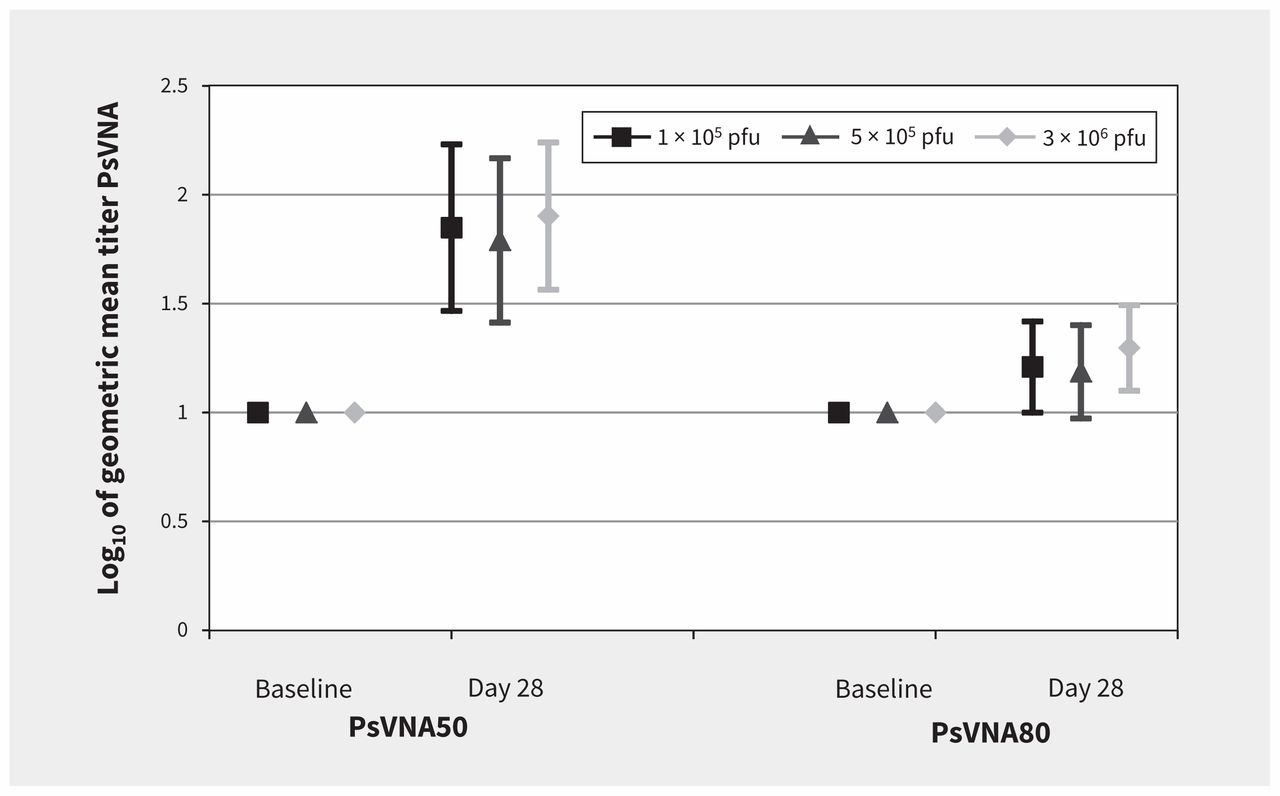
Seroconversion by day 28 using PsVNA50 was noted in 8 of 10 (80%) volunteers in the 1 × 105 and 5 × 105 pfu groups, and in 7 of 10 (70%) of the volunteers in the 3 × 106 group (Figure 3). On day 28, seroconversion by PsVNA80 was less, with rates of 20% (2 of 10 volunteers) in the 1 × 105 and 5 × 105 pfu groups, and in 1 of 10 (10%) of the volunteers in the 3 × 106 group. No difference was observed at baseline between vaccine groups and placebo.
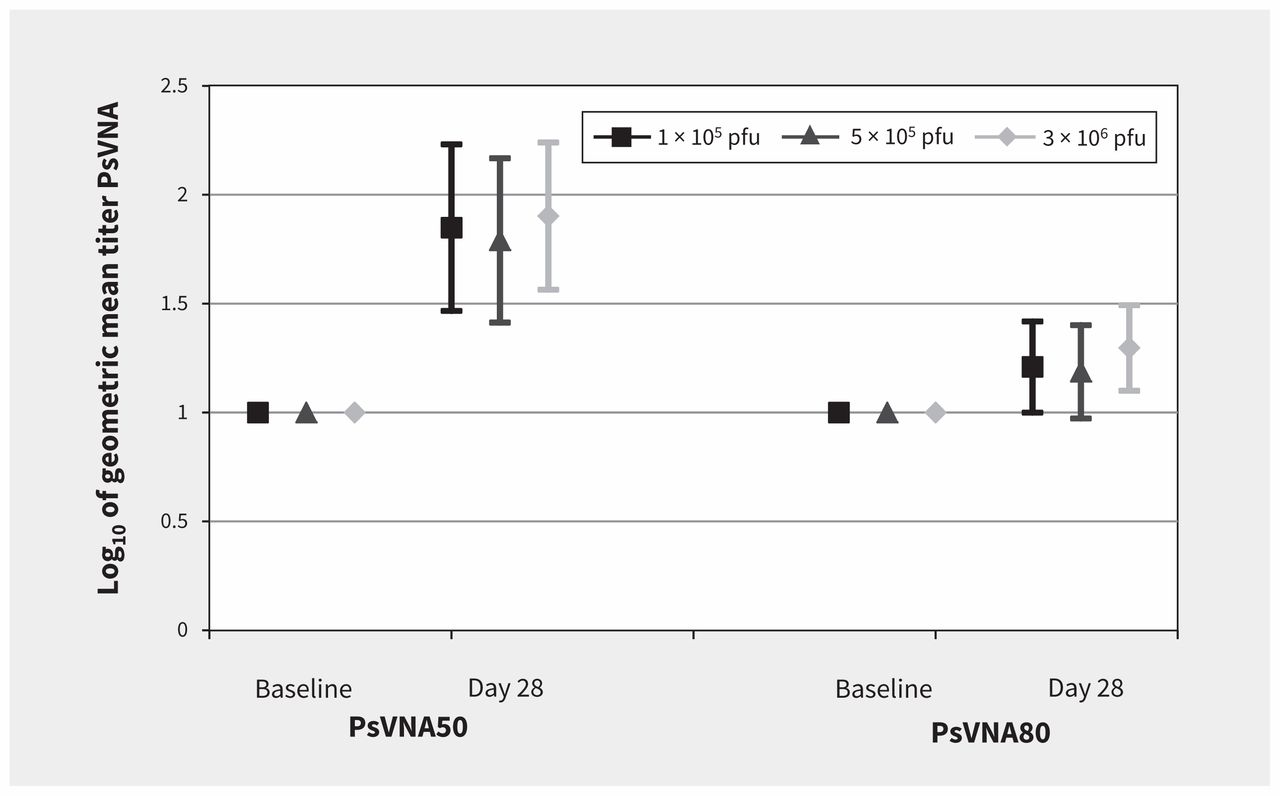
Neutralization antibody responses to Ebola glycoprotein. Note: There were no significant differences between groups using the pseudovirion neutralization assay (PsVNA) at day 28. The PsVNA50 of the 3 × 106 plaque-forming units (pfu) dose compared with the 1 × 105 pfu and 5 × 105 pfu doses was p = 0.788 and p = 0.575, respectively.
Interpretation
In this phase 1 study, all 3 dose levels of rVSVΔG-ZEBOV-GP live, attenuated vaccine were well tolerated by participants and no safety concerns were identified. Solicited AEs were primarily characterized as mild to moderate, with only 3 severe events (headache and diarrhea in the 5 × 105 pfu group; fatigue in the 3 × 106 pfu group). Arthralgia during the first 14 days post-vaccination was infrequent and not severe. Arthritis was not reported. Viremia was transient, with no detection of vaccine virus shedding in urine or saliva. In the subset of vaccinees who developed viremia, the incidence of frequently reported solicited AEs was similar or moderately increased compared with other vaccinated subjects overall. The vaccine was immunogenic, eliciting glycoprotein-binding antibodies in recipients of all 3 doses. In this report, we provide the first extended post-vaccination serology assessments and show that antibody titers persist at high levels 180 days postimmunization.
A phase 1, dose-escalation trial at WRAIR enrolled participants to assess the safety and immunogenicity of 3 × 106 pfu and 2 × 107 pfu doses.16 At the United States National Institutes of Health (NIH) Clinical Center, a 2-dose regimen was evaluated using these 2 dose levels.16 The VSV Ebola Consortium25 conducted 4 parallel phase 1 trials at sites within Europe and Africa.24 Three of the 4 trials were open label, uncontrolled and dose escalating, designed to assess the safety, AEs and immunogenicity of rVSVΔG-ZEBOV-GP doses ranging from 3 × 105 to 2 × 107 pfu. The fourth trial, in Geneva, initially administered the 1 × 107 pfu and 5 × 107 pfu vaccine doses, but was put on hold as a result of a 25% (13 of 51) and 22% (11 of 51) incidence of fever and oligoarthritis with these 2 doses, respectively. After preliminary data indicated tolerability and immunogenicity of low vaccine doses in other studies, the study was resumed in Geneva, but at a lower dose of 3 × 105 pfu, comparing outcomes to those of vaccinees who had received the higher doses before the study was put on hold.26
Patterns of detectable viremia were comparable with other studies that investigated the rVSVΔG-ZEBOV-GP vaccine; viremia was transient, peaking by day 3 and no longer evident on day 7.16,24,26 Glycoprotein-binding serum antibodies were detectable in all vaccinated individuals, irrespective of vaccine dose. Although neutralizing antibody titers were detected among most participants of our study, a dose response was not evident. A significant dose response for neutralizing antibody titers was not observed in the WRAIR phase 1 trial comparing 3 × 106 versus 2 × 107 pfu dose, but was observed in the larger VSV Ebola Consortium studies comparing multiple doses between 3 × 105 and 5 × 107 pfu dose with higher vaccine doses.16,24,26 Unlike higher doses of the rVSVΔG-ZEBOV-GP vaccine,24 the dose levels tested in our study were minimally reactogenic, which confirms the tolerability of low-dose rVSVΔG-ZEBOV-GP vaccination.16,24,26 There were no reports of arthritis among our cohort for the 180-day follow-up period, similar to published trials from WRAIR and NIH.16 Reports of arthritis with overlapping and higher doses of the vaccine suggest an intrinsic association with rVSVΔG-ZEBOV-GP, and not only a dose-dependent one.24,26
The multiple phase 1 rVSVΔG-ZEBOV-GP trials across North America (United States and Canada), Europe (Switzerland and Germany) and nonepidemic regions of Africa (Gabon and Kenya) have shown that the vaccine is well tolerated and immunogenic, and warrants further study. The range of doses administered in this study were assessed along with those in the WRAIR and NIH studies and together used to select the optimum dose to be evaluated in the phase 2/3 trials. Currently, there are 4 ongoing phase 2/3 studies: a phase 2 placebo-controlled trial in Liberia, sponsored by the US NIH; a phase 3 ring vaccination trial in Guinea, sponsored by the World Health Organization; a phase 3 trial in Sierra Leone, sponsored by the US Centers for Disease Control and Prevention; and a phase 3 immunogenicity, safety and lot consistency trial in North America and Europe, sponsored by Merck. As part of an African–Canadian collaboration, preparations for a phase 2 trial evaluating the safety and immunogenicity of rVSVΔG-ZEBOV-GP in HIV-infected adults and adolescents are underway with the intent to begin enrolling in 2017.
There are several candidate Ebola vaccines at different stages of development and testing.27 The rVSVΔG-ZEBOV-GP vaccine is the first to progress to phase 3 trials in Africa after data from the 8 phase 1 studies, including this one, collectively determined that 2 × 107 pfu is a favourable dose. Other vaccines include the Ad26-EBOV and MVA-EBOV by Johnson & Johnson and Bavarian Nordic, which have entered phase 2 and 3 trials. The recombinant protein Ebola vaccine by Novavax and Monovalent Ebola Zaire Vaccine (rVSVN4CT1-EBOVGP1) by Profectus BioSciences Inc. are being assessed in phase 1 trials. The replication-defective chimpanzee adenovirus 3 (ChAd3) is completing phase 2 studies.28–30
Limitations
Limitations of this study are primarily the small sample size, overall and per group, and the fact that only participants from North America were enrolled. Another limitation is the lack of follow-up after six months for long-term immunogenicity data.
Conclusion
Ebola transmission in Africa is now considered to be under control.2,31 However, recent clusters of cases are still being reported as a result of virus spread from shedding survivors, and more are anticipated, with the virus still present in sub-Saharan Africa’s ecosystem.32 These facts underscore the importance of continuing efforts and collaborations that may ultimately lead to licensed Ebola vaccines that would protect humans and prevent or control outbreaks in the future. Our study confirms the immunogenicity and tolerability of the rVSVΔG-ZEBOV-GP vaccine, and provides new information about duration of the antibody response.
Acknowledgements
The authors would like to thank Canadian Center for Vaccinology staff; Marilyn Tiller, IWK Research Pharmacist; and Brian Martin, Julianne Creager, Rick Nichols, Joan Fusco, Charles Link and Nick Vahanian from NewLink Genetics.
Footnotes
CMAJ Podcasts: author interview at https://soundcloud.com/cmajpodcasts/170074-res
This article has been peer reviewed.
Contributors: All authors approved the final version of this manuscript for publication and agreed to be accountable. All authors either drafted and/or revised the work. All authors contributed to one or more of the following: i) conception/design of the work; ii) data acquisition; iii) data analysis; iv) data interpretation.
Disclaimer: The opinions, interpretations, conclusions and recommendations contained herein are those of the authors and are not necessarily endorsed by the US Department of Defense.
Competing interests: May ElSherif reports grants from the Canadian Institutes of Health Research/Public Health Agency of Canada, during the conduct of the study. Donna MacKinnon-Cameron reports an operating grant from the Canadian Institutes of Health Research/Public Health Agency of Canada, during the conduct of the study. Li Li reports an operating grant from the Canadian Institutes of Health Research/Public Health Agency of Canada, during the conduct of the study. Judie Alimonti reports personal fees from NewLink Genetics, outside the submitted work; and Her Majesty the Queen in right of Canada holds a patent related to the rVSVΔG-ZEBOV-GP vaccine (patents: EU 1 527 087; US 8 012 489; CA 2,493,142). W. Jay Ramsey was an employee of NewLink Genetics during the conduct of the study, and reports personal fees as a salaried employee of NewLink Genetics, outside the submitted work. Donald Gray Heppner reports grants from the Department of Health and Human Services, during the conduct of the study and employment by NewLink Genetics Inc. (which is developing the vaccine described in this article), outside the submitted work. Tracy Kemp was an employee of NewLink Genetics during the conduct of the study; she also reports personal fees as a salaried employee of NewLink Genetics, outside the submitted work. Thomas Monath reports a grant from BARDA and the US Department of Health and Human Services, and financial support for study, personal salary and benefits from New-Link Genetics. Shelly McNeil reports an operating grant from the Canadian Institutes of Health Research/Public Health Agency of Canada, during the conduct of the study; and grants and personal fees from vaccine manufacturers (such as Merck, GlaxoSmithKline Biologicals, Sanofi Pasteur and Pfizer) outside the submitted work. Joanne Langley reports an operating grant from the Canadian Institutes of Health Research/Public Health Agency of Canada, during the conduct of the study, as well as grants and personal fees from vaccine manufacturers (such as Merck, Glaxo-SmithKline Biologicals, Sanofi Pasteur, Pfizer, and Novartis) to conduct vaccine clinical trials, outside the submitted work. Scott Halperin reports an operating grant from the Canadian Institutes of Health Research/Public Health Agency of Canada, during the conduct of the study, and grants and personal fees from vaccine manufacturers (such as Merck, GlaxoSmithKline Biologicals, Sanofi Pasteur, Pfizer and Novartis) outside the submitted work. No other competing interests were declared.
Funding: Funding was provided by the Public Health Agency of Canada and the Canadian Institutes of Health Research through a grant to the Canadian Immunization Research Network (CIRN), Funding Reference Number FRN#138434, and administered by Dalhousie University. Funding was also provided by the US Department of Defense Joint Program Executive Office for Chemical and Biological Defense Medical Countermeasure Systems’ Joint Vaccine Acquisition Program (MCS-JVAP) under contracts to the US Army Medical Research Institute of Infectious Disease (USAMRIID) and Battelle Biomedical Research Center. This research was supported in part by an appointment to the Postgraduate Research Participation Program at the USAMRIID, administered by the Oak Ridge Institute for Science and Education through an inter-agency agreement between the US Department of Energy and US Army Medical Research and Materiel Command.
Data sharing: Data from this study are the property of the Dalhousie University on behalf of the Canadian Immunization Research Network through agreements with funding partners. Data are available to others, through direct request to Scott Halperin at the Canadian Center for Vaccinology, IWK Health Centre, Halifax, NS; email: scott.halperin{at}dal.ca. Access data will be provided by sharing tables, figures or summary listings as needed by interested parties.
- Accepted May 8, 2017.
References
In this issue

Article tools






{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Immunogenicity of rVSV{Delta}G-ZEBOV-GP Ebola vaccination in exposed and potentially exposed persons in the Democratic Republic of the Congo
- Prior vaccination with the rVSV-ZEBOV vaccine does not interfere with but improves the efficacy of postexposure antibody treatment in nonhuman primates exposed to Ebola virus
- The story of Canadas Ebola vaccine
More in this TOC Section
Similar Articles


Podcast