


In the first decades of medical genetics, both physicians and patients associated this field with uncommon diseases, relevant only to specialists and those affected. This is no longer so: our understanding of the genetics underlying susceptibility to common disorders, such as diabetes mellitus, cardiovascular disease and various cancers, is bringing genetics into not only the medical mainstream, but also the public mainstream. Moreover, molecular methods are becoming important in the diagnosis and classification of malignant disease, as well as in determining prognosis and monitoring response to therapy. This paper describes current methods of studying the changes that underlie inherited and acquired disease and concludes with discussion of a technique that is generating excitement while still in development, the use of DNA microarrays or “chips.”
How does DNA encode information?
The gene is the fundamental unit of genetic information.1 Each gene is a DNA sequence — tens to millions of nucleotide bases in length and bounded by recognized control regions — which produces one or several related proteins. Genetic information is encoded by the sequence of the bases along a single strand of DNA; the bases are therefore often regarded as the alphabet forming the words, phrases and sentences of the “genetic instruction manual.” When 2 DNA strands combine to form a double helix, the nucleotide bases of one strand associate one-to-one with those on the other (Fig. 1A); genetic information is transmitted by the geometry of pairing of the bases in this double helix. Only certain associations among the 4 different types of bases are permitted, such that there are 2 sets of pairs allowed. As a consequence, either strand alone contains the information required to rebuild the other.
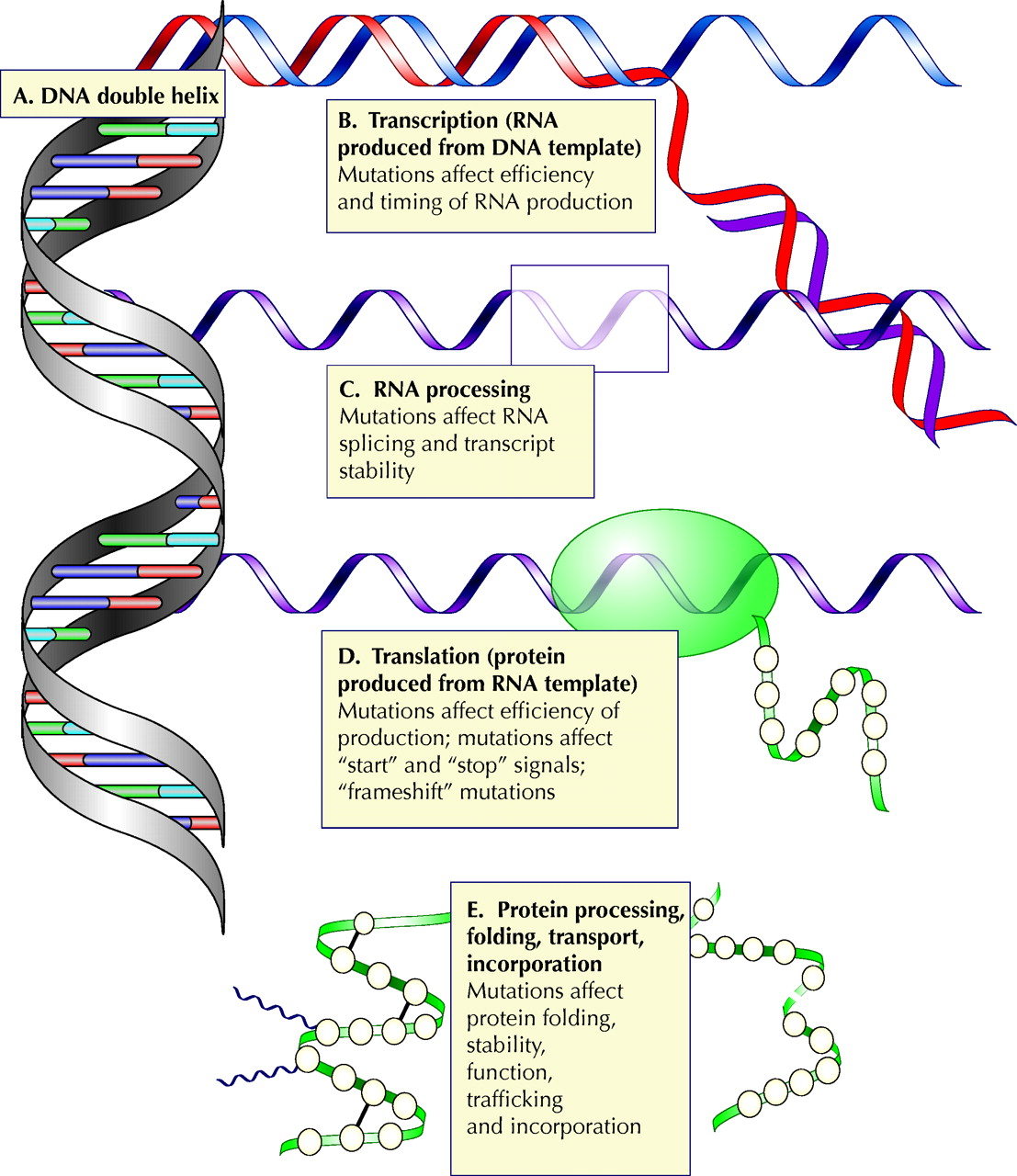
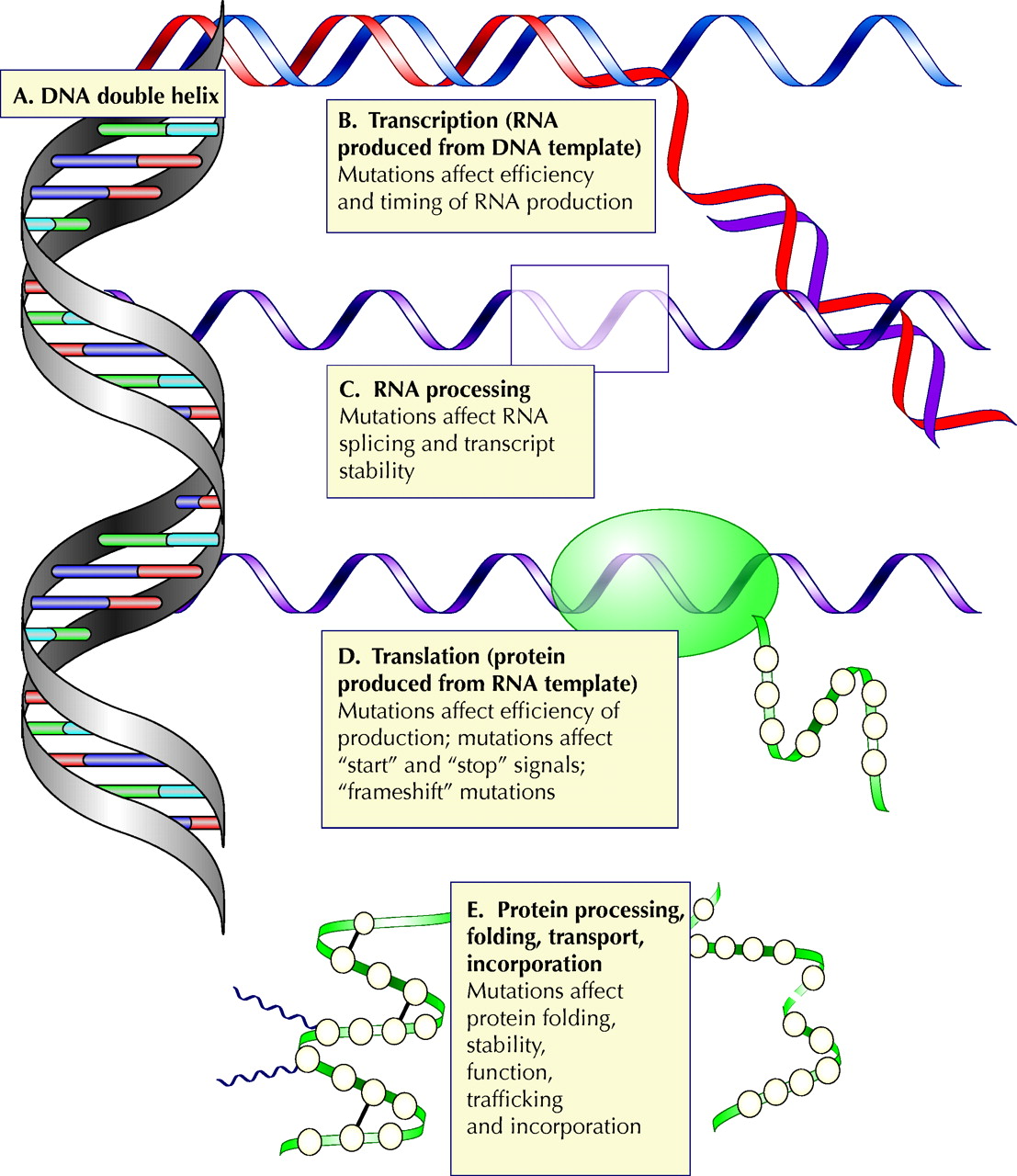
Fig. 1: Steps in the transmission of genetic information. Any of these steps may be affected by mutation. (A) Information is encoded in the DNA double helix. (B) An RNA transcript is synthesized from a DNA template. Mutations in regions controlling RNA synthesis may affect the quantity of RNA produced and may affect whether it is produced at an appropriate time in development or in the cell cycle. (C) Editing of the RNA transcript allows one mammalian gene to encode several related proteins. Mutations in sequences that control the editing may skew the relative quantities of these proteins or may cause one or more of them not to be produced. (D) RNA is translated into protein. Mutations at this stage may affect the efficiency of translation or may cause the protein to be shorter or longer than intended. (E) The protein is processed, folded and transported to its functional site in the cell. Photo: Alison Sinclair
The translation of genetic information to functional protein is a multistep process, proceeding through a transient “working copy” of RNA, which is edited to produce the template that directs the synthesis of protein. The protein is then folded into its functional form, modified further and routed to its final position in the cell (Fig. 1B–E). The process is controlled at multiple points, from control sequences, which determine when RNA will be expressed and in what quantity, to signal sequences in the protein that direct its routing.
How do mutations occur?
The “memory” of DNA does not extend beyond the current strand. If the sequence is altered (mutated) and the alteration is not corrected by the cell, subsequent replications reproduce the mutation. Mutations can arise through a variety of mechanisms and range in scale from changes to a single nucleotide to the loss, duplication or rearrangement of entire chromosomes. Certain chemicals produce DNA damage that leads to mutation, among them constituents of tobacco smoke, certain dyes and chemotherapeutic agents. Errors in the replication of DNA have been postulated as being responsible for the mutations seen in conditions such as Huntington's disease2 and myotonic dystrophy.3 Errors in recombination are responsible for mutations called translocations, such as occur in leukemias and other cancers. Normal recombination produces genetic variation by the exchange of genetic material between paired chromosomes. If recombination occurs between unpaired chromosomes, large pieces of chromosome can be inappropriately rearranged, resulting in translocations.
The effect of a mutation depends on both the mutation and its location. A single base change in the gene for an enzyme or a transporter protein may render it inactive, by changing its active site or altering its folding. Alternatively, the mutation might introduce a stop signal, causing the protein to be shortened, or remove a normal stop signal, causing the protein to be artificially elongated. Addition or removal of a single base introduces a “frame shift,” which causes misinterpretation of adjacent sequences by the protein-producing apparatus. The effects of mutations are not confined to sequences that directly encode protein: any of the multiple levels of control may be affected (Fig. 1B–E). A mutation in a control region may affect how much protein is produced (and when) and may affect whether it is produced in response to appropriate cell signals. A mutation in an RNA “splice site” (a sequence that borders RNA removed during processing) may result in inaccurate editing of the RNA transcript. A mutation in a protein signal sequence may mean that a properly folded, functional protein never reaches its final destination in the cell. Different mutations in the same gene may produce different severities of disease. For example, in β-thalassemia, the β-globin gene may be altered by deletions, nonsense mutations, splice junction mutations, frame shifts, formation of fusion genes and single base pair mutations.4 In cystic fibrosis, the most common mutation involves deletion of 3 nucleotides, which results in the protein not reaching its final destination in the membrane, but there is a plethora of additional possible mutations that alter the function of the involved chloride transporter by mechanisms that affect the protein's folding, its ability to transport chloride ions and its quantity.5,6
Larger-scale chromosomal rearrangements may result in genes for 2 different proteins being spliced together. This may produce a hybrid protein (chimera), which behaves differently from either of its progenitors, or brings a gene under the influence of different control switches, which would lead to a change in the amount and timing of its product. Translocations are often seen in cancers, some being identified as both characteristic of and prognostic for the particular tumour (e.g., the Philadelphia chromosome in chronic myelogenous leukemia and acute lymphocytic leukemia).
How are mutations identified?
Until the 1980s, the only directly observed DNA mutations were those abnormalities large enough to be detected by karyotyping, the process of examining an array of chromosomes seen under a microscope. Mutations that resulted in missing or altered protein function, such as phenylketonuria, could be inferred from the results of clinical biochemistry. Since then, innovations in the manipulation of DNA have given rise to a panoply of methods for detecting genetic abnormalities, the appropriate method depending upon the size and nature of the mutation. Some techniques are applied to the chromosomal DNA itself, some to the RNA copies produced by transcription of active DNA and some to the protein product of the gene. All exploit one or more of the basic properties of DNA or the enzymes that act upon it.
Single base pair mutations can be identified by any of the following methods:
· Direct sequencing, which involves identifying each individual base pair, in sequence, and comparing the sequence to that of the normal gene. This tends to be a labour-intensive method reserved for previously unidentified mutations or rare mutations of a common disease (such as cystic fibrosis), when other methods do not detect the disease that is clinically suspected.
· DNA hybridization methods, which make use of the strong binding of a lone DNA strand to a strand whose sequence is the perfect complement of the first and which can thus discriminate between DNA that contains mutations and DNA that contains the normal sequence. The 31 most common cystic fibrosis mutations (accounting for over 90% of the white population with this condition)6 can be identified by a hybridization-based assay.
· Restriction enzyme digestion or hybridization. Restriction enzymes are specialized enzymes that recognize and cut the DNA double helix wherever they encounter a specific very short sequence (the particular sequence depending upon the enzyme).7 Single base pair mutations may remove or, alternatively, create one of these sequences and thus alter the sizes of the DNA fragments that result.8
Larger mutations involve the deletion, rearrangement, expansion or duplication of parts of genes, entire genes or multiple genes:
· The presence or absence of a gene or substantial part of a gene can be determined by hybridization, with a labelled “probe” containing the gene sequence. If the gene is present, there will be a signal; if not, there is no signal.
· By changing the distance between sites or removing sites entirely, large mutations alter the characteristic pattern of cuts in the genome that are made by restriction enzymes. For example, the hereditary neuropathy Charcot-Marie-Tooth disease type 1A is caused by duplication of a gene encoding one of the myelin membrane proteins. Diagnosis involves characterizing the pattern produced by restriction enzymes cutting the mutated chromosome and comparing it with the normal chromosome.
· A number of strategies use the polymerase chain reaction (described in a forthcoming article9) to amplify specifically the region involving the mutation.
· Some assays detect not the mutation itself, but the altered protein it produces. Current assays for BRCA1 and BRCA2 mutations are based on detection of the truncated (shortened and nonfunctional) protein produced by the mutated genes.10
What are the limitations of laboratory testing?
As with any laboratory method, these techniques have limitations, the most important of which may be uncertainty. Although the longest-studied area of molecular medicine concerns single-gene genetic diseases, for many such diseases not all of the disease-causing genes are known. Where the genes are known, not all of the mutations have been identified, and when the potential mutations are many, as for cystic fibrosis (for which there are nearly 1000), it is not feasible to test for all of them. Nor are all mutations equally easy to identify.11 Furthermore, it is becoming increasingly apparent that even so-called single-gene disorders are products of the interaction between multiple genes.4 As a result, routine genetic testing may fail to identify mutations in 10%–40% of patients tested, even for disorders such as Duchenne's muscular dystrophy and cystic fibrosis, for which genetic diagnosis is well established.12 For less well characterized genes, the percentages are worse. The uncertainties become even more critical when genetic information is used to screen asymptomatic carriers, as these people may be basing essential life and reproductive decisions on the results.
Is it possible to test for many mutations at once?
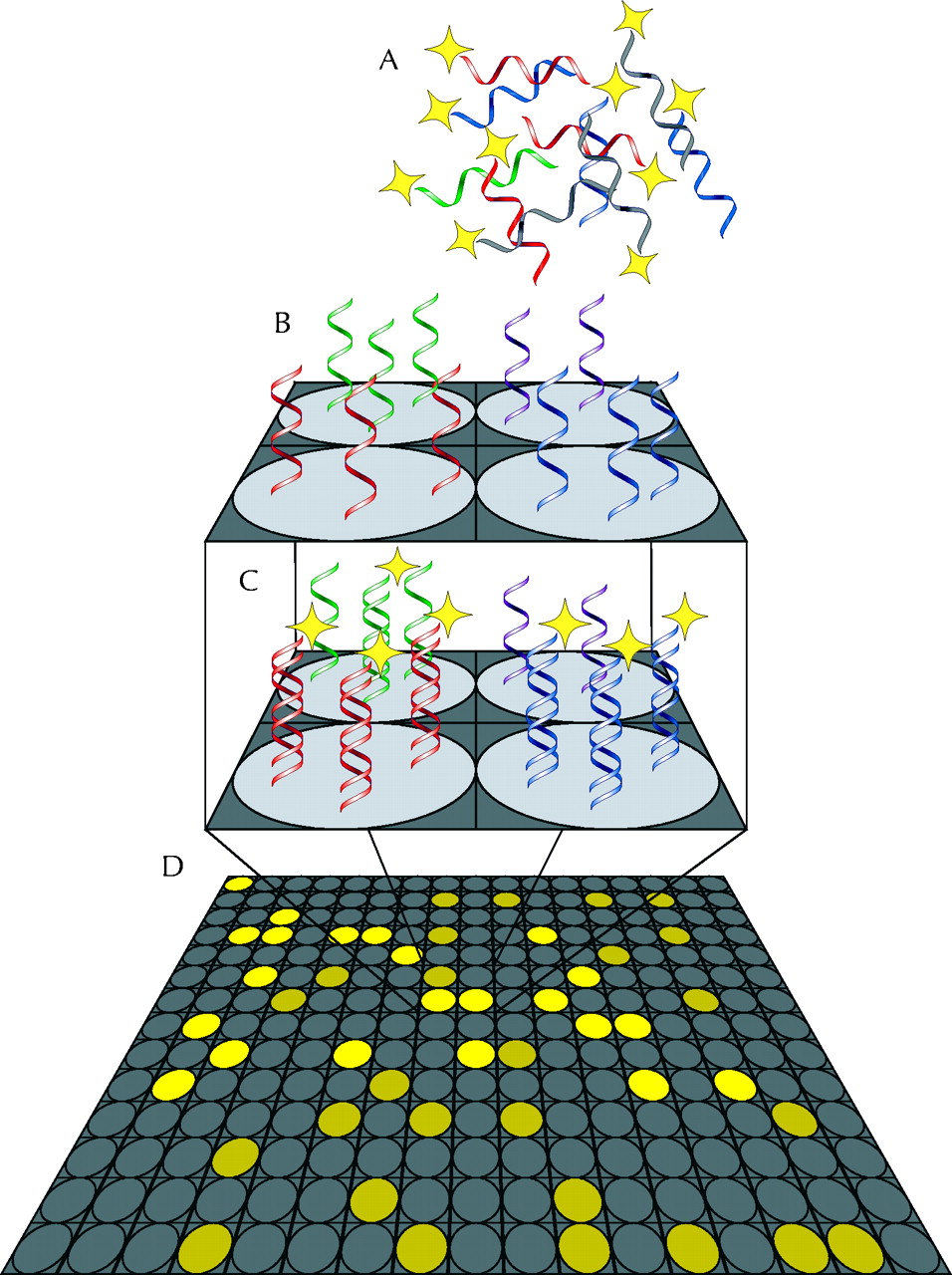
DNA “chips” or microarrays are emerging as a possible solution to the problem of testing for multiple mutations. In a DNA chip, single DNA strands containing sequences of different human genes are fixed to a solid support (such as a glass slide) in an array pattern, one gene per array point. Denatured DNA prepared from a biopsy or laboratory specimen and prelabelled with fluorescent tags is then applied to the chip (Fig. 2). Similarly, DNA that represents all the active genes in a cell can be prepared by (enzymatically) making a DNA copy of the messenger RNA in the cell. During incubation at an appropriate temperature, any DNA in the sample that is complementary to DNA on the chip binds to the fixed DNA strands that are its match. A laser is then used to detect the presence of fluorescence: a signal at any position on the array indicates that DNA for the gene fixed to the chip at that position is to be found in the sample.
{kind=link}
{kind=link}
Fig. 2: Schematic of a DNA microarray. (A) A mixture of DNA or RNA from a diagnostic sample is labelled with a fluorescent probe and layered onto a chip. (B) The chip consists of a solid support spotted in a grid pattern with thousands to tens of thousands of different single-stranded DNA sequences. (C) During incubation under carefully chosen conditions, only sequences complementary to each individual probe bind to that spot on the array. (D) When the array is read, the pattern of fluorescence indicates the sequences present in the sample and their quantities. Photo: Alison Sinclair
The power of this method is in the number of genes that can be examined simultaneously. Present-day chips can hold portions of tens of thousands of individual genes — enough to represent all the genes active in a particular cell, a lymphocyte, for instance. Chips carrying the entire set of human genes are close to realization.
Clinical application of DNA chips is still in the experimental stage. Nevertheless, in the recent literature, the pattern of gene expression observed by microarray analysis has been used to discriminate between subtypes of leukemia, lymphoma, melanoma and breast cancer, with particular emphasis on patterns of gene expression that mark tumour progression and treatment resistance.13 The genes involved with these phenomena need not have been identified; useful information can be obtained from pattern-matching alone.14
Other potential applications include genetic screening, particularly for diseases that can arise as a result of multiple possible mutations within one gene. Related applications are human genotyping, to examine multiple disease markers simultaneously to determine disease susceptibility,15 or the emerging field of pharmacogenomics, to predict responses to medications resulting from variation in types of target receptors or in enzymes used in the uptake or metabolism of drugs.16,17 In infectious disease, DNA chips might be used for the rapid identification and subtyping of bacteria, viruses and parasites, including antibiotic resistance.18,19,20,21
There are barriers to immediate wide application of the technique.22,23 The equipment is expensive, as are the arrays themselves, which are produced on a commercial scale by only a limited number of manufacturers. Relatively large amounts of DNA are used for analysis, so an amplification step is required, which must in turn amplify all sequences in the DNA proportionately. Substantial computing power is required for the analysis of thousands of data points, and there are statistical pitfalls in the analysis of a large number of data points collected from a small number of samples. In addition, gene-level examination is liable to reveal previously unappreciated variations in gene expression between normal individuals and wide heterogeneity in disease, which will lead to challenges in identifying the limits of “normal” and defining disease. However, given the strong interest in the method and its potential power, none of these barriers should prove insurmountable.
Footnotes
-
Competing interests: None declared.
Correspondence: Dr. Alison Sinclair, 406-337 St. James St., Victoria BC V8V 1J7; fax 250 380-6717; alisonsinclair{at}shaw.ca
References
In this issue
Article tools



Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles

