


- © 2005 Canadian Medical Association or its licensors
Abstract
Familial hypercholesterolemia (FH) is characterized by the accumulation of excess cholesterol in tissues including the artery wall and tendons. We describe a patient with homozygous FH who presented with asymptomatic cholesterol granuloma of the brain. The patient's plasma low-density lipoprotein cholesterol level was remarkably responsive to combination hypolipidemic therapy with statin plus ezetimibe. This case illustrates another potential complication of whole-body cholesterol excess and underscores the differences in phenotype and in response to therapy among patients with FH.
A 45-year-old Chinese-Canadian woman presented with chemosis of several years' duration. As a teenager the patient had noticed corneal arcus and thickening of her Achilles tendons. At age 20 she was found to have a fasting total cholesterol level of 18.0 (normal < 5.2) mmol/L and a low-density lipoprotein (LDL) cholesterol level of 15.4 (normal < 3.4) mmol/L. On the basis of these findings and a family history of hypercholesterolemia in both parents and premature coronary artery disease in her father, homozygous familial hypercholesterolemia (FH) was diagnosed. The initial treatment was with bile acid-binding resins, followed by the addition of hydroxymethylglutaryl coenzyme A reductase inhibitors, or statins, when she was 29. She has been intolerant of fibrates and niacin, both of which caused rashes. At age 30, angina pectoris necessitated coronary artery bypass grafting, and she has since remained free of cardiovascular symptoms. At age 40, carotid ultrasound imaging, conducted after carotid bruits were detected, revealed more than 95% and 50%–79% stenoses of the left and right carotid arteries respectively. She began taking 325 mg of ASA daily and has remained free of neurologic symptoms. Her total cholesterol level and LDL cholesterol level fell to 10.1 and 8.2 mmol/L respectively after she began taking 80 mg of atorvastatin and 4 g of colestipol daily, and her Achilles tendon xanthomas also shrank over time. Her lipoprotein (a) level was elevated at 0.44 (normal < 0.3) g/L. Her sitosterol level was 2.4 (normal ≤ 5.0) μg/mL, and her homocysteine level was 8.0 (normal < 12.1) μmol/L); her ApoE genotype (ε3/ε3) was also normal. The patient's high-density lipoprotein cholesterol level was 0.8 (normal > 0.9) mmol/L on no lipid therapy and has ranged between 0.6 and 1.1 mmol/L with atorvastatin and colestipol therapy; her triglyceride level was 2.7 (normal < 2.3) mmol/L at baseline and has ranged from 1.4 to 3.6 mmol/L with therapy.
A genomic DNA sequence analysis showed that she was homozygous for proline-to-leucine substitution at residue 644 of the LDL receptor known as FH-Zambia1 or FH-Gujerat,2which has not been reported in Canada.3 This mutation is associated with slowed maturation and attenuated cell-surface expression of LDL receptors4 but does not abolish receptor function. Partial LDL receptor function appears to explain the patient's responsiveness to therapy, which was better than expected for most patients with homozygous FH. The addition of 10 mg daily of ezetimibe to the atorvastatin and colestipol therapy lowered the patient's LDL cholesterol level to 6.9 mmol/L. When colestipol was discontinued, this level decreased further to 5.9 mmol/L, which suggests an enhanced response to combination statin–ezetimibe therapy in the absence of bile acid-binding resin. Fig. 1 shows the sites of action of medications taken by the patient. We are considering LDL apheresis to further decrease LDL levels.
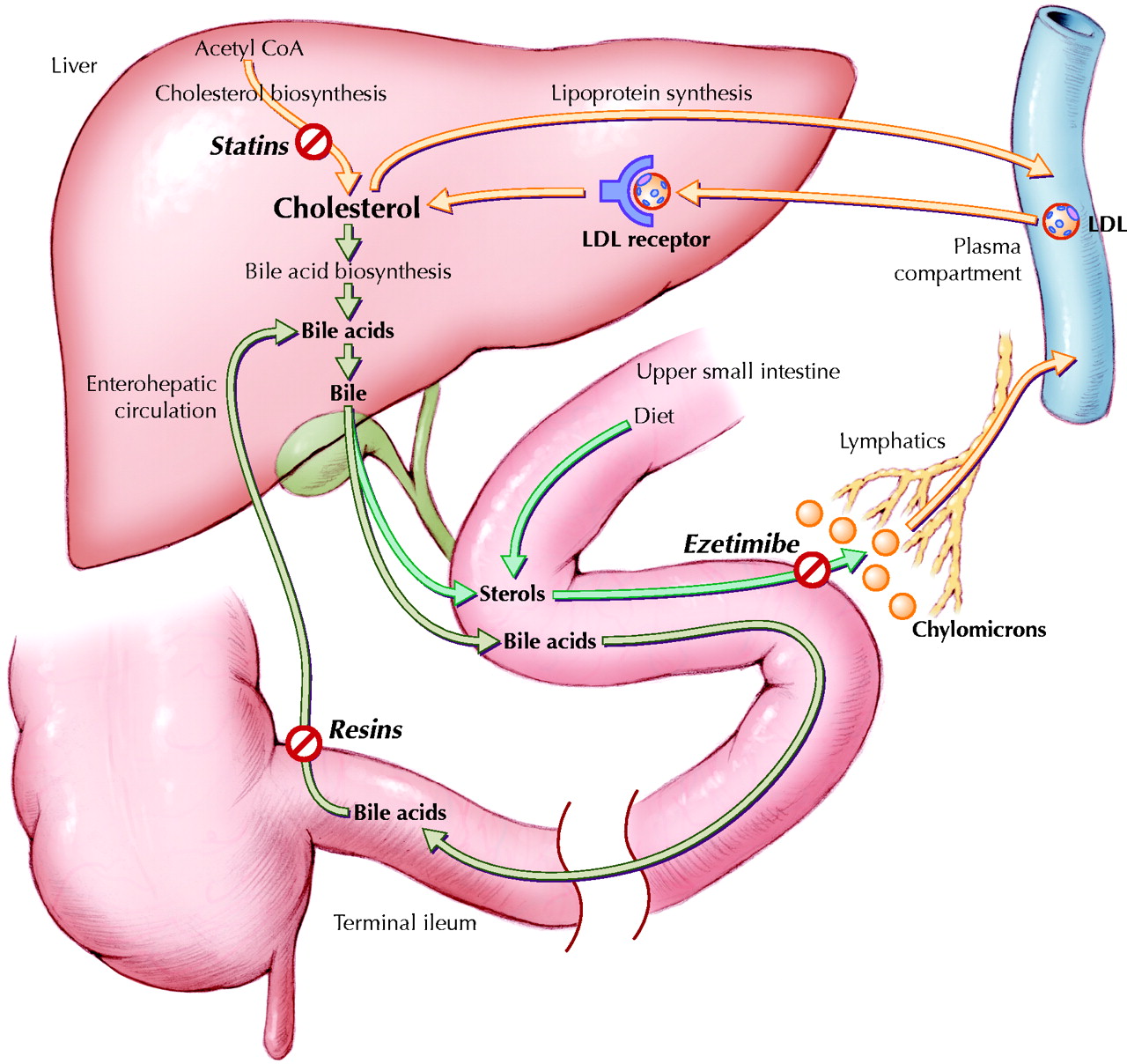
Fig. 1: Sites of action of medications taken by the patient. The liver is central to cholesterol metabolism. Hepatic cholesterol can be synthesized from acetyl coenzyme A (CoA) in a multistep enzymatic process, whose rate-limiting enzyme (3-hydroxy-3-methylglutaryl CoA reductase) is inhibited by statins such as atorvastatin. Hepatic cholesterol is used in part to synthesize bile acids that are destined, with sterols, for secretion in bile. The luminal sterol pool in the upper portion of the small intestine comes from both dietary and biliary sterols. Intestinal luminal sterols are transported into enterocytes and repackaged into chylomicrons for secretion into lymph and, ultimately, plasma. Ezetimibe likely inhibits sterol absorption by interfering with the sterol transporter Niemann–Pick C1 Like 1 protein.5 Bile acids advance through the intestinal lumen and facilitate intestinal cholesterol absorption. Unbound bile acids are normally recycled through the terminal ileum into the enterohepatic circulation. Bile acid-binding resins, such as colestipol, prevent this recycling and force more hepatic cholesterol into bile acid synthesis. To compensate for the depletion of hepatic cholesterol stores induced by all of these drugs, the liver increases expression of cell-surface low-density lipoprotein (LDL) receptors to extract more cholesterol from the plasma by receptor-mediated endocytosis. Although both copies of the LDL-receptor gene were defective in this patient — causing very high baseline plasma LDL cholesterol — each had some residual activity, and she was able to respond to treatment. See the animated figure at www.cmaj.ca/cgi/content/full/172/4/495/DC1. Photo: Lianne Friesen and Nicholas Woolridge
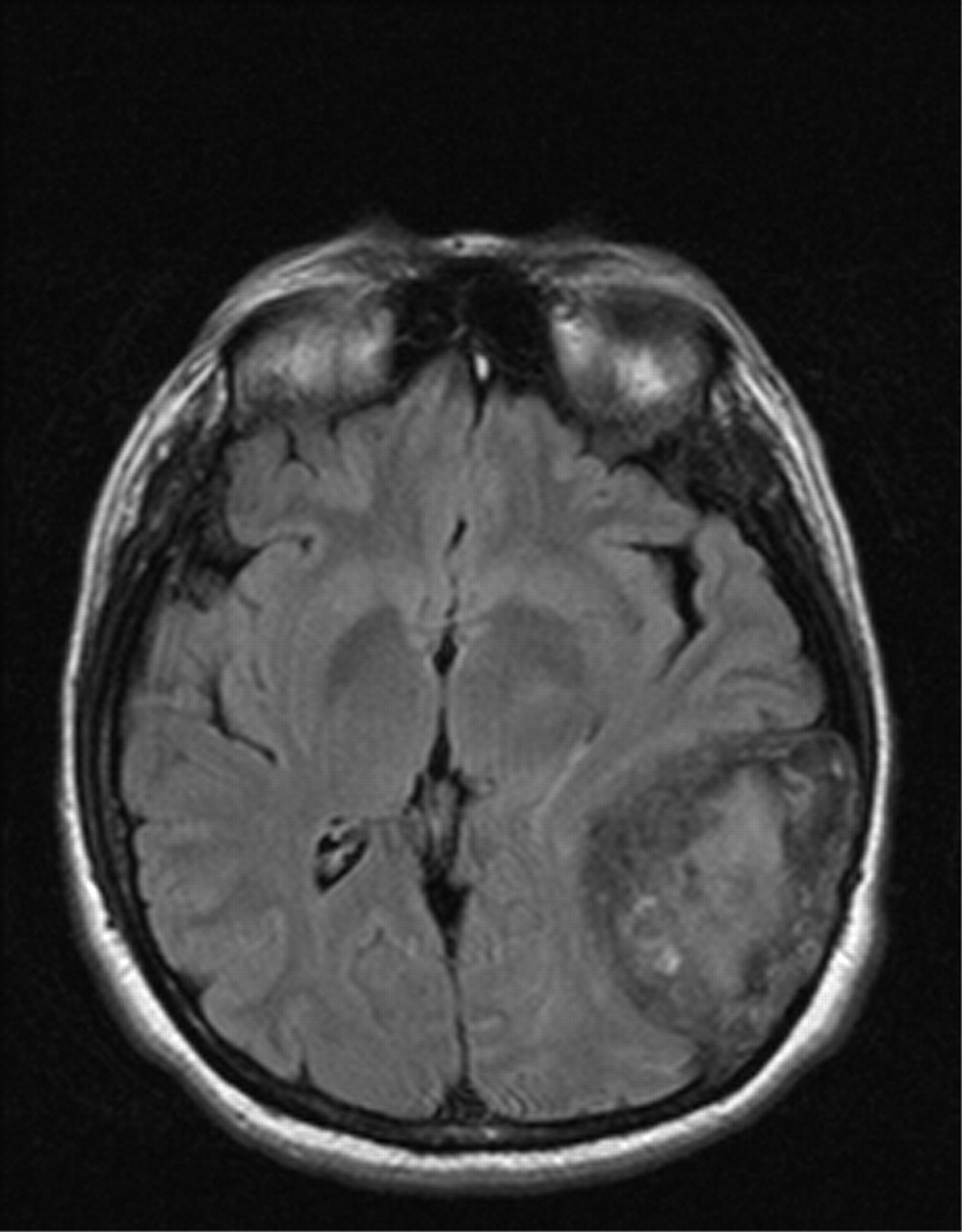
Ophthalmologic examination revealed bilateral chemosis and conjunctival redness with no ocular hypertension or congested retinal vasculature. The patient was clinically and biochemically euthyroid, with no thyroid antibodies. Although the chemosis was felt to have resulted from chronic use of vasoconstricting eye whiteners, a head MRI scan was performed to rule out thyroid ophthalmopathy. It revealed an incidental large calcified mass involving the skull and meninges of the left temporal-occipital area (Fig. 2). Craniotomy for the resection of a presumed meningioma revealed a large, encapsulated, avascular mass emanating from the petrous bone that was filled with necrotic tan-to-yellowish paste-like material. Microscopy of the lesion's capsule showed cholesterol clefts with granulomatous tissue including lipid-containing macrophages, multinucleated giant cells, chronic inflammation and fibrosis, with the contents showing shards of cholesterol and foci of calcification, consistent with a cholesterol granuloma. The patient had no perioperative complications or neurologic sequelae. A head CT scan performed 18 months after the operation showed no residual or recurrent mass, which is typical following resection of these tumours if the petrous bone is properly pneumatized.6 The chemosis persists, but we feel that, rather than being related to the tumour, it is a consequence of rebound vasodilatation secondary to chronic use of eye whiteners, even though the patient has stopped using them.

Fig. 2: MRI of a large intracranial cholesterol granuloma complicating homozygous familial hypercholesterolemia.
Most cerebral cholesterol granulomata have not been associated with genetic hypercholesterolemia,7,8 with only 1 previous case linked to homozygous FH.9 These lesions arise silently and are thought to occur as a foreign body reaction to cholesterol crystals deposited in or adjacent to the mastoid air cells following blockage of the air cell system, in which negative pressure and hypoxia lead to local tissue and blood cell necrosis.7 Cholesterol granulomata occur in about 1 per 2–3 million population each year.8,10 They frequently cause neurosensory hearing loss or cranial nerve palsies or both,7,8 but our patient had neither. She also had no history of known antecedents to cholesterol granulomata, including cholesteatoma, otitis media or cerebral bleeding.7 It seems surprising that cholesterol granulomata have not been more commonly reported in cases of homozygous FH, a condition with whole-body cholesterol excess.1 The absence of neurologic symptoms despite the large intracranial mass was likely a result of the very slow growth of the granuloma. The constellation of an asymptomatic cerebral cholesterol granuloma, comparatively late-onset cardiovascular disease and relatively good response to hypolipidemic therapy underscores the phenotypic heterogeneity among patients with homozygous FH. Cerebral cholesterol granulomata should be added to the list of potential complications of FH.
Footnotes
-
This article has been peer reviewed.
Contributors: Gordon Francis, Royce Johnson and Max Findlay provided medical care to the patient, and Jian Wang and Robert Hegele performed the genomic DNA sequence analysis. Gordon Francis and Robert Hegele wrote most of the article. All authors contributed to the revision of the article and approved the final version.
Competing interests: Royce Johnson, Max Findlay and Jian Wang have no conflicts of interest to report. Gordon Francis and Robert Hegele have received consultancy fees and speakers honoraria from Pfizer, Merck Frosst/Schering and AstraZeneca.
In this issue

{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
More in this TOC Section
Similar Articles
Collections

