


Bullous hemorrhagic dermatosis is an uncommon nonimmune adverse drug reaction to heparin-based anticoagulants including unfractionated heparin, low-molecular-weight heparin and fondaparinux that typically occurs within 7 days of drug exposure.
The reaction is more common in older adults, and lesions are usually found on the extremities.
There are typically no changes to coagulation tests such as the international normalized ratio, partial thromboplastin time or the prothrombin time.
Bullous hemorrhagic dermatosis is self-resolving, although careful monitoring of bleeding, and supportive management such as wound care and transfusion of blood products, may be required.
A 90-year-old woman was admitted to a tertiary hospital with hypoxia secondary to pulmonary edema and ongoing antibiotic therapy following an infected hip prosthesis. The patient’s medical history included heparin-induced thrombocytopenia following administration of dalteparin, atrial fibrillation (CHADS2 score of 3) and lower gastrointestinal bleeding related to diverticular disease. The patient did not report any skin allergies. On admission, she was 4 weeks post-removal of right hip hardware, débridement and revision to a cemented right hip hemiarthroplasty for methicillin-sensitive Staphylococcus aureus infection, and was completing a 6-week course of cephalexin and rifampin. She had previously taken apixaban for stroke prophylaxis but, before admission, her apixaban was switched to warfarin because of its interaction with rifampin. However, given the interaction between warfarin and rifampin and her complex history of thrombosis, we consulted the thrombosis service regarding management of the patient’s anticoagulation. We then began daily fondaparinux injections into her abdomen while continuing rifampin. The patient’s dyspnea resolved with intravenous furosemide.
On the fourth day of fondaparinux, the patient developed deep purple hemorrhagic lesions not associated with trauma — which were neither painful nor pruritic — and bullae on her left forearm (Figure 1). These progressed in size and number over the next 2 days, eventually affecting her right arm but remained isolated to the upper extremities. Mucosal membranes were not involved. The left epicondylar bullae ruptured on day 6 of fondaparinux resulting in substantial hemorrhage. While the patient’s hemoglobin dropped from 91 (normal 130–180) g/L on day 4 of fondaparinux therapy to 70 g/L on day 6, her coagulation parameters were in the upper normal range: international normalized ratio (INR) 1.2 (normal 0.8–1.2), prothrombin time (PT) 14.9 (normal 11–15) seconds, and partial thromboplastin time 41 (normal 22–35) seconds. We did not identify any other source of bleeding or hemolysis. We gave the patient 4 units of packed red blood cells over 3 days, and discontinued fondaparinux on day 7 because of ongoing bleeding. We dressed her skin lesions with tranexamic acid–soaked compression dressings. A thorough medication review did not show other potential causes of hemorrhagic bullae.
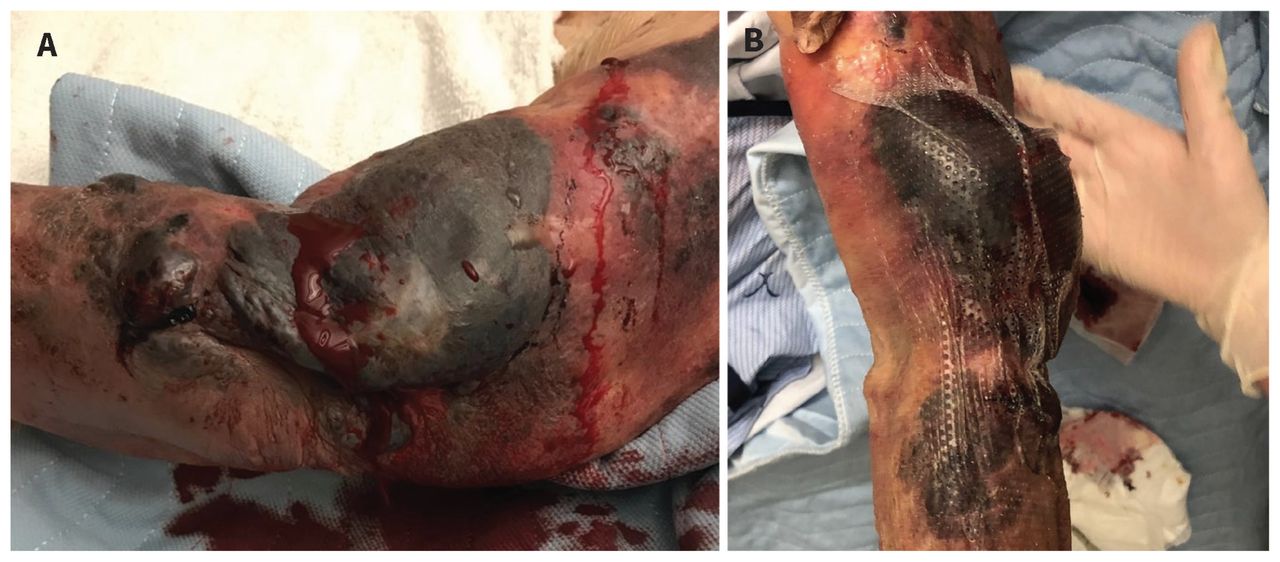
Photographs of bullous hemorrhagic dermatosis lesions on the left arm and hand of a 90-year-old woman, on day 6 (A) and day 10 (B) of fondaparinux administration.
Rheumatologic panels on day 6 showed a positive extractable antibody panel with positive anti-Ro/SSA52 at 4.1 (normal < 1.0). We suspected vasculitis-related hemorrhagic bullae, but a punch biopsy from the left arm taken on day 10 showed only dermal solar elastosis and atrophic epidermis, which was not consistent with vasculitis. In consultation with the rheumatology service, we decided that the extractable antibody panel result was likely spurious.
We diagnosed fondaparinux-induced bullous hemorrhagic dermatosis. We clearly documented possible life-threatening reactions to all heparin-related products on her medical chart, and discussed this with the patient and her family.
On the advice of the infectious diseases team, we discontinued rifampin and restarted the patient on low-dose apixaban for ongoing stroke prophylaxis. The hemorrhagic bullae completely resolved after we discontinued fondaparinux, with desiccation of the bullae and involution of the skin discoloration on day 23 (Figure 2).

Resolution of bullous hemorrhagic dermatosis on the patient’s left arm on day 23.
This hospital admission profoundly affected our patient’s quality of life. Owing to deconditioning and resultant frailty she ultimately applied to long-term care.
Discussion
Epidemiology and differential diagnosis
Bullous hemorrhagic dermatosis is an uncommon, non-immune-mediated, systemic adverse effect of heparin-based anticoagulants. It has been most commonly reported with enoxaparin, and typically appears after 7 days (range 3–270 d) of medication exposure.1,2 Bullous hemorrhagic dermatosis is likely underreported, and only 97 cases have been described in peer-reviewed journals.1,3–6 The condition has been reported previously in relation to a combination of fondaparinux and warfarin.7,8 There have been no reported cases associated with direct oral anticoagulants. In a summary of 91 reported cases of bullous hemorrhagic dermatosis, Russo and colleagues found that lesions are not typically painful, pruritic or associated with other systemic symptoms; and lesion distribution is most often distant to injection sites, with primary sites affected being the extremities, followed by the torso, with rare involvement of the mucosal membranes.1,4 In most cases, bullous hemorrhagic dermatosis is self-resolving, with time to resolution within an average of 13 days.9 Four deaths have been associated with the condition.1
The range of differential diagnoses for hemorrhagic bullae is broad (Table 1). For our patient, the differential included warfarin-induced skin necrosis, vasculitis-related hemorrhagic bullae, cephalexin-induced bullous pemphigoid, mechanical friction blisters, acquired hemophilia and bullous hemorrhagic cellulitis. The onset of warfarin-induced skin necrosis is typically within a few days of drug exposure and occurs in areas with subcutaneous fat, including breast and thighs. This did not fit the onset or distribution of lesions in our patient. Bullous pemphigoid and many forms of drug-mediated cutaneous reactions are immune-mediated responses associated with eosinophil infiltration, which was not evident on our patient’s skin biopsy.7,10
Hemorrhagic bullae related to an acquired hemophilia (e.g., acquired factor VIII deficiency) are also immune related, and would be expected to change anticoagulation parameters such as international normalized ratio or partial thromboplastin time.11 There was also no evidence of skin or systemic infection. Given the punch biopsy pathology report, the time correlation with the onset of fondaparinux, self-resolution of the lesions and a lack of plausible alternative explanations, we diagnosed a probable case of fondaparinux-induced bullous hemorrhagic dermatosis according to the World Health Organization–The Uppsala Monitoring Centre causality assessment system.12
Pathophysiology
The pathophysiology of bullous hemorrhagic dermatosis has not been fully elucidated. Skin biopsies tend to show intra-epidermal hemorrhage in the absence of immune cells or markers of inflammation.1 Given the advanced average age of patients with bullous hemorrhagic dermatosis, some have theorized a relationship to epidermal-dermal fragility. This would be consistent with our patient’s case, given the atrophic epidermis described in her skin biopsy. She had crepe skin consistent for her advanced age but no prior noted skin lesions or breakdown.
Our patient may have been predisposed to bullous hemorrhagic dermatosis because of a combination of increased risk of skin necrosis from heparin-induced thrombocytopenia 6 months previously, delayed skin healing and increased skin fragility owing to age and comorbidities.1 Heparin-induced thrombocytopenia is an immunoglobulin G antibody–mediated adverse drug reaction to heparin products.13 Case reports have described a possible association between bullous hemorrhagic dermatosis and eczematous reactions at injection sites, suggesting a type IV hypersensitivity reaction.1 This is a different immune mechanism from that involved in heparin-induced thrombocytopenia and does not confer susceptibility to other immune reactions. There have been no reported cases of increased venous or arterial thromboembolism in patients with bullous hemorrhagic dermatosis, which is central to the diagnosis of heparin-induced thrombocytopenia. Given that bullous hemorrhagic dermatosis is not immune mediated, the patient’s history of heparin-induced thrombocytopenia and subsequent development of bullous hemorrhagic dermatosis with fondaparinux are likely coincidental.
Management
Stopping the offending agent or transitioning to an alternative anticoagulation agent are the most common treatment strategies, and have been used in about 60% of reported cases.1 Given that bullous hemorrhagic dermatosis tends to be self-limited, some experts have argued that stopping the offending agent makes little difference in time to resolution. About 23% of cases resolved even though the suspected medication causing bullous hemorrhagic dermatosis was continued, and 14.3% resolved on a lowered dose or change in anticoagulant.1 Given the severity of our patient’s case, which included hemodynamic instability requiring transfusion, we considered that discontinuing all anticoagulation agents was prudent. We have reported this case to the Canada Vigilance Program.
Conclusion
Bullous hemorrhagic dermatosis is a rare nonimmune, self-resolving drug reaction that occurs with heparin-based anticoagulants. This reaction is more common in older adults, and can result in hemodynamic instability requiring transfusion. The risks and benefits of ongoing anticoagulation must be evaluated on an individual basis.
The section Cases presents brief case reports that convey clear, practical lessons. Preference is given to common presentations of important rare conditions, and important unusual presentations of common problems. Articles start with a case presentation (500 words maximum), and a discussion of the underlying condition follows (1000 words maximum). Visual elements (e.g., tables of the differential diagnosis, clinical features or diagnostic approach) are encouraged. Consent from patients for publication of their story is a necessity. See information for authors at www.cmaj.ca.
Footnotes
Competing interests: None declared.
This article has been peer reviewed.
The authors have obtained patient consent.
Contributors: All of the authors contributed to the conception and design of the work, drafted the manuscript, revised it critically for important intellectual content, gave final approval of the version to be published and agreed to be accountable for all aspects of the work.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is noncommercial (i.e., research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/
In this issue

{kind=link}
{kind=link}
Article tools






Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Collections

