


Understanding of the pathophysiology of thrombotic microangiopathies — a group of rare yet life-threatening hematologic conditions — has evolved in recent years along with better access to diagnostic testing. Identifying secondary causes requires a thorough diagnostic workup, but appropriate treatment of the underlying condition usually leads to resolution.
We review evidence on the pathophysiology, diagnosis and management of this group of conditions for the generalist (Box 1). We summarize disease mechanisms and highlight controversies surrounding diagnosis and treatment of thrombotic microangiopathies, starting with a review of disease terminology.
Evidence used in this review
We searched MEDLINE, Embase and conference abstracts for our evidence review. Search terms included “hemolytic uremic syndrome,” “microangiopathic hemolytic anemia,” “thrombotic microangiopathy,” “thrombotic thrombocytopenic purpura,” their abbreviations (i.e., HUS, MAHA, TMA, TTP, respectively), and terms related to their pathophysiology, diagnosis and management. We assessed primary research and relevant review articles and reviewed their reference lists.
What are thrombotic microangiopathies?
Thrombotic microangiopathies are a group of disorders characterized by microangiopathic hemolytic anemia, thrombocytopenia and microthrombi leading to ischemic tissue injury.1 Though rare, thrombotic microangiopathies are life-threatening conditions that require urgent management. Presenting symptoms may be nonspecific, but basic laboratory tests reveal a specific constellation of thrombocytopenia and anemia with red blood cell fragmentation on the blood film and evidence of hemolysis. Primary care or emergency department physicians are often the first point of contact; however, once the diagnosis is suspected, urgent referral is needed to a specialty service (e.g., hematology or nephrology) that can provide plasma exchange.
Microangiopathic hemolytic anemia is the hallmark of thrombotic microangiopathy. It is a process of red blood cell destruction within the microvasculature accompanied by thrombocytopenia due to platelet activation and consumption. Thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS) are primary forms of thrombotic microangiopathies. The incidence of TTP in adults is about 3 per 1 000 000,2 and the incidence of HUS in children is about 3 per 100 000.3 Regardless of the etiology, thrombotic microangiopathy is a hematologic emergency that requires prompt treatment.
In recent years, our understanding of pathophysiology has improved considerably. Once thought to be interchangeable terms, TTP and HUS have distinctly different mechanisms.4–6 Thrombotic thrombocytopenic purpura is caused by a severe deficiency of the enzyme ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) due to acquired autoantibodies or, rarely, genetic mutations. The most common form of HUS (typical HUS) follows a diarrheal illness caused by Shiga toxin–producing Escherichia coli; whereas atypical HUS is associated with abnormal host susceptibility to complement-mediated damage.5 These advancements in understanding have also led to confusion about disease classification, in particular with respect to the role of ADAMTS13 testing.
How are thrombotic microangiopathies classified?
Thrombotic microangiopathy is the overarching term used to describe any disease process characterized by thrombocytopenia and microangiopathic hemolytic anemia with or without other clinical or laboratory features. These disorders can be divided into primary or secondary forms. Primary thrombotic microangiopathies (TTP and HUS) occur spontaneously with no associated underlying cause. Secondary forms occur in the context of pregnancy, autoimmune disease, malignancy, bone marrow transplantation or use of certain medications, and treatment should be aimed at correcting the underlying cause (Table 1).7,27
Differential diagnoses of thrombotic microangiopathies and diagnostic investigations
Thrombotic thrombocytopenic purpura is broadly defined as a thrombotic microangiopathy occurring in the context of severe ADAMTS13 deficiency (< 10%).7 ADAMTS13 is a normal enzyme in plasma that cleaves large forms of the coagulation protein von Willebrand factor into smaller functional subunits. The acquired form of TTP is thought to be due to an autoantibody against ADAMTS13, and the inherited form is due to a genetic deficiency of ADAMTS13. In 1996, Furlan and colleagues28 and Tsai29 independently found that patients with TTP had unusually large von Willebrand factor multimers in their plasma and that this was due to a reduction in the levels of the cleaving enzyme. Large von Willebrand factor multimers bind to platelets and form platelet-rich fibrin strands, which leads to intravascular hemolysis and ischemic tissue injury. Tsai and Lian10 and Furlan and colleagues11 subsequently identified inhibitory antibodies as the cause for acquired disease. In 2001, the von Willebrand factor cleaving enzyme was identified as ADAMTS13 based on kindred studies involving patients with the congenital form of TTP called Upshaw–Schulman syndrome.8 Congenital TTP often presents in childhood but can manifest for the first time after a “second hit” later in life; for women, this can occur during or immediately after pregnancy.8,9
Hemolytic uremic syndrome is a thrombotic microangiopathy associated with severe renal impairment and normal or slightly reduced ADAMTS13 activity. The most common form (typical HUS) is associated with bloody diarrhea due to Shiga toxin–producing E. coli.12 The diagnosis requires identification of the Shiga toxin or the enterohemorrhagic strain of E. coli from stool. Atypical HUS is less common. It is caused by a dysregulation of the alternative complement pathway, which leads to increased complement activity.13 About 50%–70% of patients with atypical HUS have genetic mutations in complement regulatory genes30 and a small subset of patients have autoantibodies against complement Factor H, a major regulator of the alternative complement pathway.16
How is the diagnosis of thrombotic microangiopathy made?
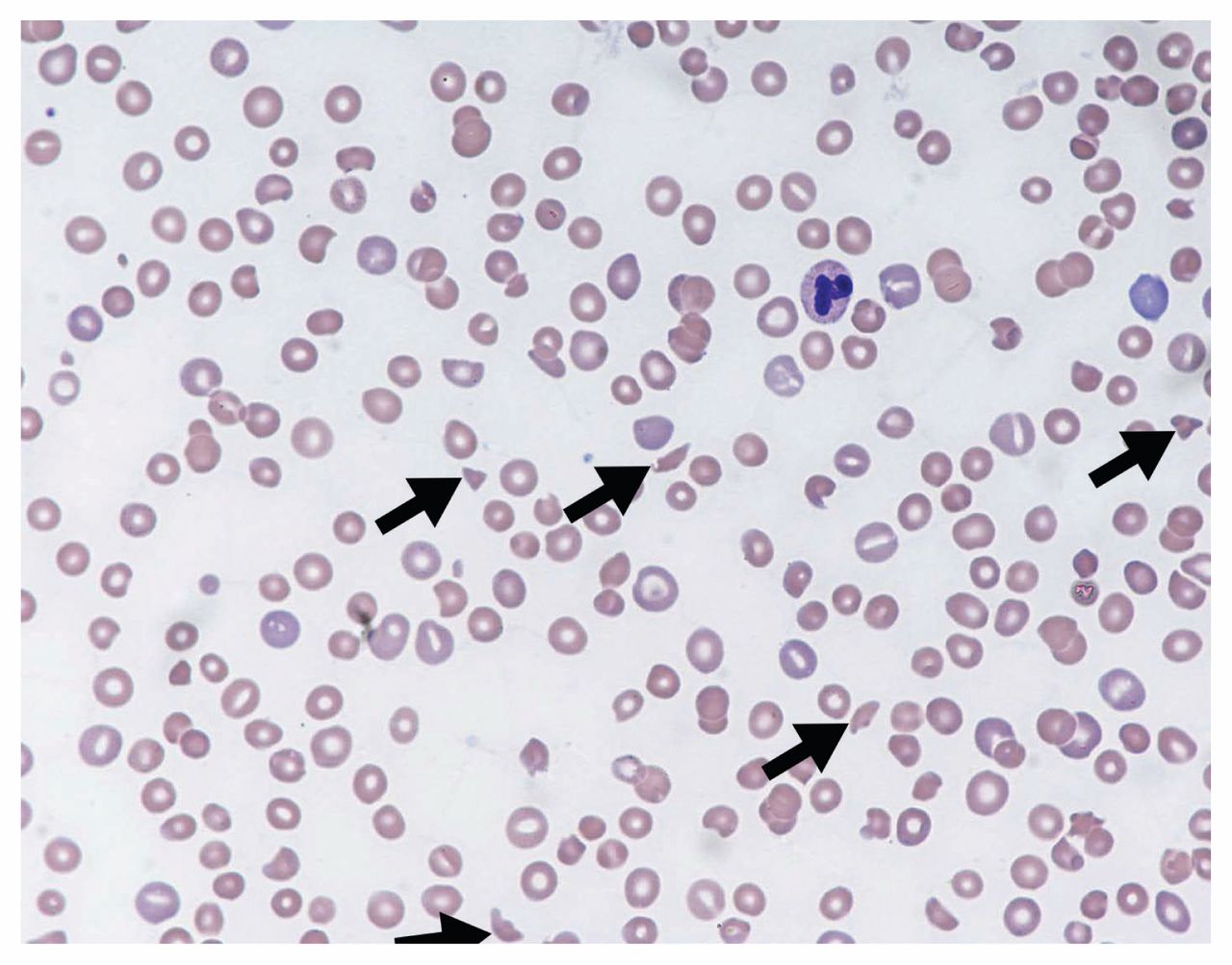
Thrombotic microangiopathy is a clinicopathologic diagnosis.1 The constellation of thrombocytopenia, anemia and red blood cell fragmentation (i.e., schistocytes) on the blood film is sufficient to make the diagnosis (Figure 1). The finding of concomitant anemia and thrombocytopenia should prompt a request (through direct communication with the laboratory staff) for a peripheral blood film to look for red blood cell fragmentation. In the appropriate clinical context, more than two schistocytes per high-powered field31 or more than 1.0% schistocytes32 on a blood film is considered pathologic; however, these cut-offs have not been validated.

Peripheral blood film showing classic findings of thrombotic microangiopathy. The image shows features of thrombocytopenia and microangiopathic hemolysis (Wright–Giemsa stain, original magnification × 50). Arrows show red blood cell fragments (i.e., schistocytes), which are substantially reduced. Image courtesy of Dr. Catherine Ross (Juravinski Hospital & Cancer Centre, McMaster University, Hamilton, Ont.).
For TTP, the full pentad of signs and symptoms (thrombocytopenia, schistocytic anemia, neurologic impairment, renal impairment and fever) is present in only 5% of patients.31 The first two criteria — thrombocytopenia and schistocytic anemia — are enough to initiate treatment, especially since early mortality is high with about 50% of deaths occurring in the first 24 hours.33
Other laboratory abnormalities in patients with thrombotic microangiopathy are reticulocytosis, elevated lactate dehydrogenase, unconjugated hyperbilirubinemia and low haptoglobin. The direct antiglobulin test is usually negative, and coagulation tests (international normalized ratio and partial thromboplastin time) are normal but may become elevated in patients with very severe disease associated with disseminated intravascular coagulation. Kidney biopsy can aid in identifying thrombotic microangiopathy as the cause of renal impairment, but typical histologic findings (e.g., arteriolar or capillary thickening, endothelial edema or detachment, fibrin or platelet-rich thrombi) are nonspecific.1
Once thrombotic microangiopathy is suspected, a thorough search for secondary causes is important to help guide treatment. This should include a pregnancy test for females of childbearing potential and careful questioning about drug exposures, HIV risk factors and other infections. ADAMTS13 testing should be done before the institution of plasma therapy to avoid false-negative results; however, this should not delay treatment. Severe ADAMTS13 activity (< 10%) is indicative of TTP.7,34
Stool cultures or polymerase chain reaction testing for Shiga toxin should be done if there is a preceding diarrheal illness in order to identify patients with typical HUS.12 If no secondary cause is identified and ADAMTS13 activity is not severely reduced, complement mutation analysis should be requested to identify patients with atypical HUS.35
Monitoring of patients with thrombotic microangiopathy should include serial testing of serum creatinine, urinalysis for protein, electrocardiography, troponin levels and liver enzymes.7,31
What is the initial treatment for thrombotic microangiopathies?
Thrombotic thrombocytopenic purpura is a hematologic emergency. Untreated, it is associated with mortality as high as 90%; with plasma exchange, mortality can be reduced to 20%.4 Once TTP is suspected, plasma exchange should be instituted promptly. Plasma exchange is a procedure in which plasma from the patient is removed and replaced with fresh donor plasma by use of an apheresis machine. If plasma exchange is not immediately available, plasma infusion can be administered while waiting for plasma exchange to be arranged or for the patient to be transferred to a facility that can provide it.
Evidence supporting the use of plasma exchange derives from a Canadian randomized trial involving patients with TTP, which showed improved response (47.1% v. 25.5%, p = 0.025) and reduced mortality (3.9% v. 25.5%, p = 0.035) compared with plasma infusion.4 Clinicians should avoid treatment delays and misdiagnoses by not waiting to base treatment decisions on results of ADAMTS13 testing. Folic acid supplementation should be administered to avoid folate depletion from ongoing hemolysis. 7 Platelet transfusions should be reserved only for patients with serious bleeding related to thrombocytopenia36 because this treatment has been associated with increased thrombosis.37
Corticosteroids are frequently added to plasma exchange as part of initial therapy.31 The typical regimen is prednisone 1 mg/kg orally followed by a rapid taper over three to four weeks once a normal platelet count has been achieved. Higher doses may be used for patients with more severe disease. One randomized trial of high- or standard-dose methylprednisolone (n = 60) showed improved remission rates by day 23 in the high-dose group (77% v. 47%, p = 0.03).38 In our experience, some patients with TTP will respond to plasma exchange alone; thus, corticosteroids may not be required initially but can be added if the platelet count, hemolytic markers and clinical symptoms do not improve after one or two days.
Antiplatelet agents might prevent ongoing platelet aggregation and minimize microthrombus formation. In one randomized trial from 1997, the use of acetylsalicylic acid plus dipyridamole followed by ticlopidine maintenance for one year showed a trend toward improved survival in patients with acute TTP.39 Other studies did not show such a benefit. The use of antiplatelet agents for patients with thrombotic microangiopathy is controversial but might be reasonable as secondary prevention in patients with a platelet count greater than 50 × 109/L.39
Hemolytic uremic syndrome associated with Shiga toxin– producing E. coli usually resolves with supportive care (e.g., fluid rehydration, red cell transfusions).40 Antimicrobial treatment may worsen the disease, depending on the bacterial strain and antibiotic class.41 Plasma exchange is rarely needed.42
For patients with atypical HUS, plasma exchange should be administered initially because the clinical features are often indistinguishable from TTP. The diagnosis of atypical HUS should be considered in patients with substantial renal impairment who do not respond to plasma exchange and who do not have severe ADAMTS13 deficiency.14 For those patients, eculizumab, a mono-clonal antibody against the terminal complement component C5, should be considered.5,43 In a prospective phase 2 study involving two cohorts (n = 17, trial 1; n = 20, trial 2), 88% and 80% achieved thrombotic microangiopathy event-free status, respectively, by the end of the 26-week trial; after two years, event-free status was seen in 88% and 95%, respectively.44 A single-arm study (n = 41 adults with atypical HUS) reported that 73% of patients achieved remission after treatment with eculizumab and 79% were able to stop receiving dialysis. Two patients developed meningococcal infection, a recognized risk with terminal complement inhibition.43
What is the treatment for relapsed or refractory thrombotic microangiopathy?
Refractory TTP is generally defined as the lack of rise in platelet count, persistent hemolysis, lack of symptom resolution or new neurologic signs after five to seven days of plasma exchange.45 Relapse is defined as disease recurrence after remission, usually 30 days after the last plasma exchange treatment.45,46 Evidence from a retrospective study suggests that relapsed or refractory TTP can be managed successfully by increasing the intensity or frequency of plasma exchanges47 and adding or increasing doses of immunosuppressant therapy, including corticosteroids.38 Rituximab has been shown to be effective in refractory TTP,46 and some investigators have used it as initial therapy.48 The proteasome inhibitor bortezomib has also been effective for some patients, although controlled trials have not been done.49,50
Other treatments include cyclosporine,51 cyclophosphamide, 52 vincristine,53 N-acetylcysteine7 and splenectomy, which can reduce the rate of relapse in patients with recurrent TTP.54 Emerging therapies include caplacizumab, a novel anti–von Willebrand factor nanobody,55 and recombinant ADAMTS13.56
For patients with relapsed or refractory atypical HUS who require renal transplantation, pre-emptive use of eculizumab can improve transplantation outcomes.57 Combined liver–kidney transplantation can restore renal function and increase the levels of complement regulatory proteins that are produced by the liver.58 Patients with refractory atypical HUS should be re-evaluated to ensure that secondary causes are not missed.
What is the role of ADAMTS13 testing?
ADAMTS13 testing has no role in the initial management of thrombotic microangiopathy. Severely reduced levels of ADAMTS13 are consistent with the diagnosis of TTP and can distinguish TTP from atypical HUS; however, results of ADAMTS13 testing should not be used to guide initial treatment. If TTP is suspected, plasma exchange should be instituted regardless of the results of ADAMTS13 testing.
The prevalence of ADAMTS13 deficiency among patients suspected of having TTP is variable, ranging from 18% to 100%.59,60 In single-centre studies, virtually all patients with TTP had ADAMTS13 deficiency,60 whereas in studies involving patients referred from other institutions, about 50% of those with suspected TTP had ADAMTS13 deficiency.61 This variability relates to case mix and overlapping clinical signs; for example, patients who present with thrombocytopenia and schistocytic anemia may have an alternative diagnosis (e.g., sepsis, malignancy) that only becomes apparent over time. In addition, many patients with secondary thrombotic microangiopathy will respond to plasma exchange.62
Other limitations of ADAMTS13 as a diagnostic test are logistical issues related to timing of the sample and interassay variability. ADAMTS13 levels can become falsely elevated because of exposure to plasma, although the effect is modest.34 Blood samples for ADAMTS13 testing should be collected before plasma therapy begins whenever possible. ADAMTS13 assays also differ in their detection methods and performance characteristics. Functional assays measure ADAMTS13 activity by incubating patient plasma with a source of von Willebrand factor and measuring the amount of residual uncleaved protein.63
Newer assays, such as the fluorescence resonance energy transfer assay, measure enzyme activity using synthetic von Willebrand factor peptides as the substrate. The use of these assays will improve reliability of detecting severe ADAMTS13 deficiencies across samples and across laboratories.64,65 Anti-ADAMTS13 autoantibodies, which can help distinguish acquired from congenital forms of TTP, are measured by determining von Willebrand factor cleavage activity after serial dilutions with donor plasma. Results are reported in Bethesda units, similar to the measurement of other coagulation factor inhibitors; however, these tests require local expertise and validation.
ADAMTS13 deficiency can be used to predict the risk of recurrence and can provide useful prognostic information. Bendapudi and colleagues66 found that patients with TTP and ADAMTS13 deficiency had better response to plasma exchange and better overall survival than those without ADAMTS13 deficiency. Patients with early recovery of enzyme activity had improved outcomes compared with those with persistent deficiency.34 For patients in remission, persistent ADAMTS13 deficiency is a risk factor for disease recurrence and in certain situations might prompt the use of plasma exchange as a preventive treatment; for example, during pregnancy9 or before major cardiac surgery.67 Figure 2 summarizes our treatment algorithm highlighting the role of ADAMTS13 testing.

Proposed diagnostic and treatment algorithm for thrombotic microangiopathy.4,7,39,44 CT = computed tomography, ECG = electrocardiography, HUS = hemolytic uremic syndrome, LDH = lactate dehydrogenase, TTP = thrombotic thrombocytopenic purpura. *Refractory TTP: Persistent thrombocytopenia and persistent elevation of LDH level despite therapy. †Exacerbation: Recurrent disease within 30 days of stopping plasma exchange. ‡Remission: Response for more than 30 days after last plasma exchange. §Relapse: Return of disease more than 30 days after stopping plasma exchange.
Unanswered questions
Several unanswered questions remain. What are the clinical and laboratory predictors of relapse in patients with TTP? What is the risk of relapse during pregnancy in women with a history of TTP? Should rituximab be used as initial treatment for all patients with acute TTP? How long should patients with atypical HUS receive treatment with eculizumab?
Conclusion
The identification of ADAMTS13 levels and corresponding inhibitory antibodies represents a major advancement in the pathophysiology of TTP, and testing has become more accessible. ADAMTS13 testing can support a diagnosis of TTP and may help identify patients at high risk for recurrent disease, but should not be used to guide initial therapy. Similarly, without a definitive diagnostic test, atypical HUS is primarily a clinical diagnosis that should be considered in patients who do not have severe ADAMTS13 deficiency and who do not respond to plasma exchange. Testing for complement abnormalities can confirm the diagnosis. Management of relapsed and refractory disease represents an important clinical challenge. Future research will require multicentre, multinational collaborations and large prospective patient registries.
KEY POINTSThrombotic microangiopathies are hematologic emergencies that require urgent intervention.
Since the full pentad of signs and symptoms (thrombocytopenia, schistocytic anemia, neurologic impairment, renal impairment and fever) occurs in only 5% of patients, treatment should not be delayed in those found to have thrombocytopenia and microangiopathic anemia alone.
Testing for severe ADAMTS13 deficiency should not delay the decision to initiate treatment with plasma exchange.
Refractory or relapsed TTP requires intensification of plasma exchange treatments and immunosuppressant medications.
Acknowledgements
The authors thank Dr. Catherine Ross for providing the blood film image. Donald Arnold holds the John G. Kelton Chair in Translational Research from McMaster University.
Footnotes
Competing interests: Christopher Patriquin is the recipient of an unrestricted educational grant from Alexion Pharmaceuticals (makers of eculizumab) and has participated in advisory boards with the company. No other competing interests were declared.
This article has been peer reviewed.
Contributors: All of the authors contributed to the conception or design of the work, drafted and revised the article, approved the final version and agreed to act as guarantors of the work.
References
In this issue

{kind=link}
{kind=link}
Article tools



Jump to section
- Article
- What are thrombotic microangiopathies?
- How are thrombotic microangiopathies classified?
- How is the diagnosis of thrombotic microangiopathy made?
- What is the initial treatment for thrombotic microangiopathies?
- What is the treatment for relapsed or refractory thrombotic microangiopathy?
- What is the role of ADAMTS13 testing?
- Unanswered questions
- Conclusion
- Acknowledgements
- Footnotes
- References
- Figures & Tables
- Related Content
- Responses
- Metrics
Related Articles
Cited By...
- Clopidogrel-induced thrombotic microangiopathy: a case report
- Dedifferentiated liposarcoma with heterologous rhabdomyosarcomatous differentiation in the bone marrow
- Persistent renal replacement requirement following fulminant psittacosis infection in pregnancy
- A systematic review of adeno-associated virus gene therapies in neurology: the need for consistent safety monitoring of a promising treatment
- Platelet Activating Immune Complexes Identified in COVID-19 Associated Coagulopathy
- Adalimumab as a potential cause of drug-induced thrombocytopaenic microangiopathy
- Thrombotic thrombocytopenic purpura masquerading as a stroke in a young man
More in this TOC Section
Similar Articles
Collections

