


- © 2007 Canadian Medical Association or its licensors
The Case: A 62-year-old woman had been admitted to hospital in July 1992 with flu-like symptoms and malaise. At that time, she had a 5-year history of hypertension and newly diagnosed diabetes mellitus. She denied having chest pain and had no pertinent family history. An electrocardiogram (ECG) obtained on admission showed ST-segment depression over leads V1 through V4 and nonpathological Q waves over V4 through V6. After admission, shock developed that required inotropic support, and treatment for a suspected non-ST-segment elevation myocardial infarction (MI) was started. This diagnosis was supported by serial cardiac enzyme levels: peak creatine kinase 592 (normal < 235) U/L and lactate dehydrogenase 383 (normal < 246) U/L. Echocardiography revealed left ventricular hypertrophy (LVH) and mild hypokinesia. Cardiac catheterization revealed concentric LVH, but normal coronary arteries. The patient was discharged a few days later, was well on follow-up and had no chest pain.
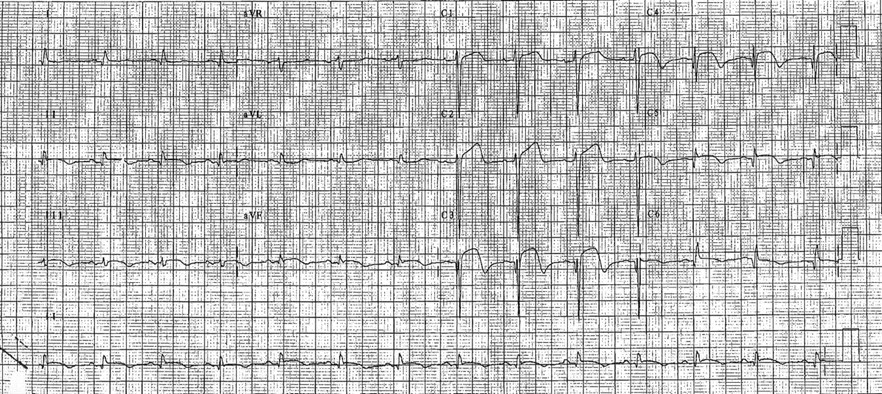
Four years later, the patient was admitted to hospital with vomiting, dizziness and chest pain. She was hypotensive on admission. An ECG showed ST-segment elevation over leads V2 through V5 (Fig. 1). An echocardiogram revealed a large anterior MI with akinesia of the apex, the distal intraventricular septum and the anterior wall. Minimal mitral valve regurgitation was noted. The patient had elevated cardiac enzyme levels and was treated for a suspected recent MI. Her condition was further complicated by episodes of hypotension, which required inotropic support. In view of a loud systolic murmur on examination, the patient had a repeat echocardiogram, which revealed an atrial thrombus, but no evidence of a ventricular septal defect. Her condition gradually stabilized, and warfarin treatment was started. She was discharged 12 days later, and the results of a post-discharge exercise tolerance test were negative.

Fig. 1: Twelve-lead ECG performed in April 1996 showing ST-segment elevation over leads V2 through V5.
Over the next 12 months, the patient was admitted on 2 other occasions because of chest discomfort and dizziness. Her cardiac enzyme levels were elevated, and an ECG showed changes on both occasions; therefore, treatment for acute coronary syndromes was started. Recurrent syncopal episodes with documented postural hypotension also developed. Warfarin therapy was stopped in August 1997 following an echocardiogram that showed no further evidence of atrial thrombus. A repeat echocardiogram in September 1997 showed marked septal left ventricular hypertrophy with a mild subaortic gradient, systolic anterior movement of the mitral valve and mid-systolic closure of the aortic valve with mild mitral valve regurgitation, which suggested hypertrophic obstructive cardiomyopathy (see video 1, available online at www.cmaj.ca/cgi/content/full/176/2/171/DC1).
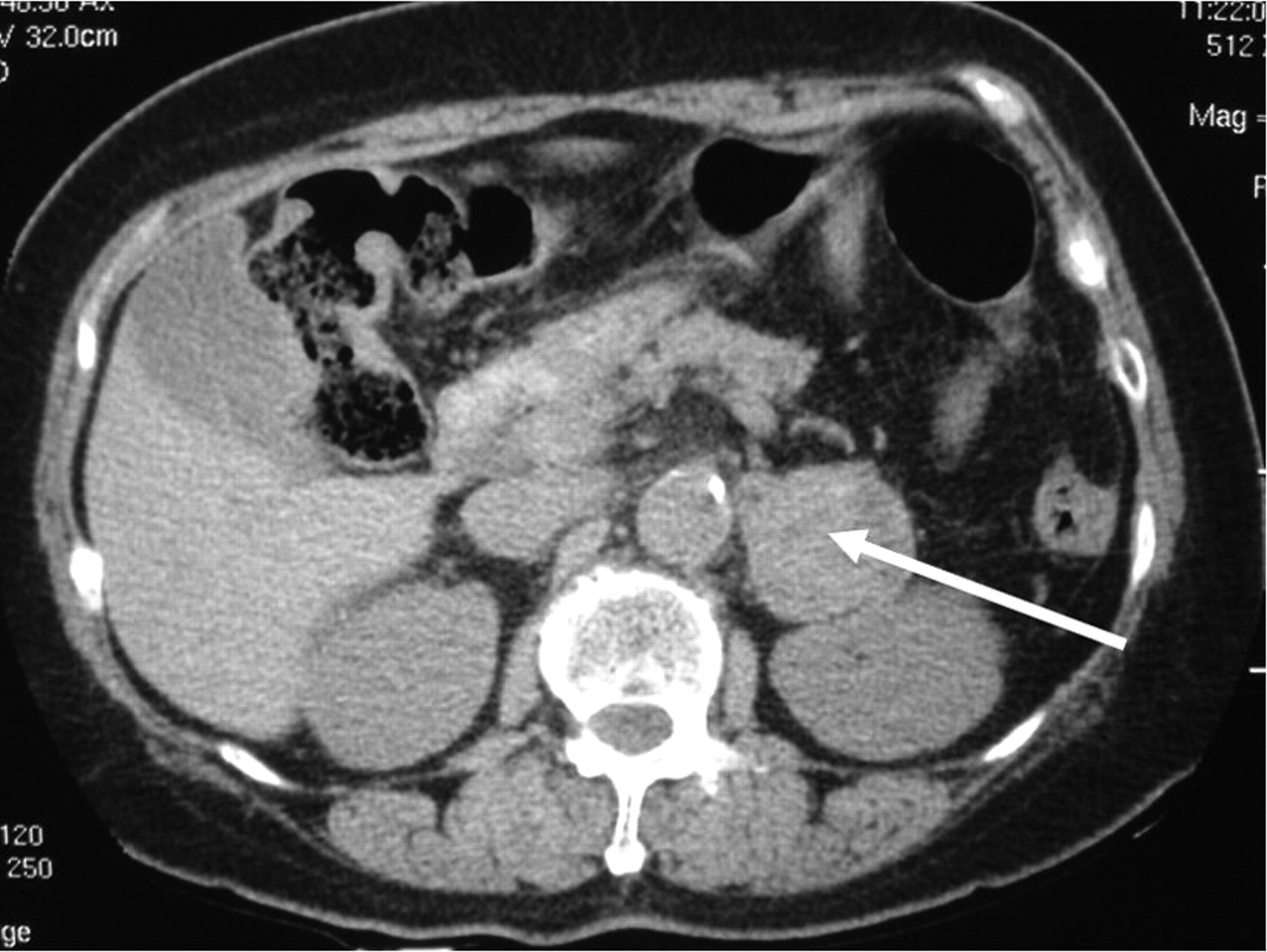
At a follow-up visit in March 1998 the patient described persistent dizzy spells and occasional headaches. She was noted to have suboptimal blood pressure control. A 24-hour urine collection for catecholamines was performed, which revealed marked elevation of catecholamine levels: epinephrine 1239 (normal 19–113) nmol/d and norepinephrine 1743 (normal 63–416) nmol/d. A CT scan revealed a 4-cm lesion on the left adrenal gland (Fig. 2). A meta-iodobenzylguanidine (MIBG) scan showed tracer uptake in the left adrenal gland consistent with the diagnosis of a pheochromoctyoma producing epinephrine and norepinephrine. Phenoxybenzamine hydrochloride and pro-pranolol therapy was begun, and the patient underwent laparoscopic adre-nalectomy in December 1998. A 4-cm hemorrhagic nodule with cystic areas was removed along with the left adrenal gland. Histology confirmed a pheochromoctyoma with no malignant features.

Fig. 2: CT scan of abdomen showing a 4-cm lesion on the left adrenal gland (arrow).
The patient's postoperative recovery was unremarkable, apart from episodes of palpitations with frequent runs of narrow complex tachycardia documented on a Holter monitor. Urinary catecholamine levels were normal, as were thyroid function test results. The patient was prescribed sotalol for arrhythmia prophylaxis. She remained well with no further episodes of chest pain or dizziness. Her diabetes resolved postoperatively: the fasting blood glucose level was 5.9 mmol/L and the hemoglobin A1c concentration was 5.8%. Her blood pressure also improved after tumour resection, and the antihypertensive medication therapy was stopped. An ECG showed only LVH, although a repeat echocardiogram in January 2004 showed a sigmoid-shaped left ventricle with marked septal hypertrophy, but normal left ventricular outflow tract with no measurable gradient. An abnormal relaxation pattern with diastolic dysfunction was noted; however, systolic function was normal (see video 2, available online at www.cmaj.ca/cgi/content/full/176/2/171/DC1). When last seen, in October 2005, the patient was well, with a blood pressure of 144/75 mm Hg, and was taking ASA, famotidine, simvastatin, sotalol and isosorbide mononitrate.
The patient we described presented with recurrent episodes of chest pain and vomiting as well as ECG changes suggestive of cardiac ischemia. Subsequent investigations, including coronary angiography, did not show evidence of substantial coronary artery disease. The patient also had marked LVH resembling hypertrophic obstructive cardiomyopathy. These cardiac complications were later found to be caused by a pheochromoctyoma. Pheochromocytoma can present with a myriad of symptoms (Box 1). This diagnosis is often not suspected, and at least 35% of pheochromocytomas are diagnosed at autopsy. If the classic symptoms of sweating, palpitations and headache are absent, as in the case we have described, it could be difficult to diagnose. Our patient was likely to have harboured the pheochromocytoma at the time of her first admission in 1992, because her long history of hypertension and diabetes was resolved after removal of the tumour.

Although patients with pheochromocytoma typically describe paroxysms, there is marked variability in its manifestations between patients that can be attributed to the effects of catecholamines. Most patients with pheochromocytoma have hypertension, which may be intermittent, remittent or persistent. Paroxysms of severe hypertension occur in about 50% of cases. After an intense and prolonged episode of hypertension, shock may ultimately occur. This may be caused by low plasma volume, arrhythmias, cardiac damage or loss of vascular tone. In addition, epinephrine secretion from a pheochromocytoma, as in the present case, can cause episodic hypotension and syncope. This is attributed to concomitant stimulation of β2 receptors, which results in vasodilatation in skeletal muscles. Excess catecholamine levels can also precipitate arrhythmias, which were present in about 20% of patients with catecholamine-secreting tumours in one study.1
Pheochromocytoma can induce myocardial damage in a variety of ways. Long-standing hypertension can cause ventricular hypertrophy and, as in this case, marked septal hypertrophy. Coupled with intravascular volume depletion and impaired diastolic filling, outflow tract obstructions simulating hypertrophic obstructive cardiomyopathy may occur. In such cases, the echocardiographic features have been noted to improve following tumour resection.2 In addition, persistent and prolonged exposure to high levels of catecholamines can result in dilated cardiomyopathy or catecholamine myocarditis, which is characterized by foci of myocardial cellular necrosis. In rare instances, pheochromocytoma has been reported to mimic an acute myocardial infarction. In these cases, there may be marked ECG changes, including T-wave inversion and ST-segment elevation.3 Catecholamines can alter ion transport across cell membranes and can alter the rate of membrane depolarization, which results in ECG changes that are suggestive of ischemia. In addition, ECG changes may be accompanied by segmental or global myocardial dysfunction, which may lead to pulmonary edema.4 The pathophysiology of the myocardial dysfunction associated with pheochromocytoma has been linked to a direct toxic effect induced by catecholamines, demand ischemia due to increased myocardial oxygen consumption, and myocardial stunning caused by coronary spasm. In the present case, the lack of residual contractile dysfunction or ECG abnormalities after acute episodes with ischemic changes and marked left ventricular regional hypokinesia suggested myocardial stunning to be the underlying mechanism.
The diagnosis of pheochromocytoma is established by demonstrating excessive levels of catecholamines or their metabolites in blood or urine. It is now recognized that the measurement of free metanephrines in plasma or fractionated metanephrines in urine offers the highest sensitivity and specificity for the diagnosis of a pheochromocytoma.5 Imaging studies should be arranged to localize the tumour after the demonstration of unequivocal biochemical evidence of a pheochromocytoma. Definitive surgery can often be performed laparoscopically but requires careful preoperative preparation. Preoperative α-blockade can normalize ischemic changes on an ECG due to catecholamine-induced cardiac ischemia.
This unusual case illustrates several of the cardiovascular manifestations of pheochromocytoma. Pheochromocytoma should be included in the differential diagnosis of acute coronary syndromes, and clinicians should be aware of the less common presentations of pheochromocytoma in order to diagnose this potentially fatal condition (Box 2).

Footnotes
-
This article has been peer reviewed.
Competing interests: None declared.
In this issue
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools



Related Articles
Cited By...
- No citing articles found.
More in this TOC Section
Similar Articles
Collections

