


Management of patients living with amyotrophic lateral sclerosis (ALS) requires specialized multidisciplinary holistic care.
Disease-modifying pharmacologic therapies to treat ALS include riluzole and edaravone.
Close attention to nutritional support and respiratory care is required for optimal care in ALS.
Multiple treatments are available to ease the symptoms of ALS.
Palliative care and caregiver support are important components of assisting patients along their journey with ALS.
Amyotrophic lateral sclerosis (ALS) is a debilitating, progressive disease with degeneration of motor neurons in the brain and spinal cord causing weakness, muscle atrophy, fasciculations and spasticity.1 Onset in the limbs, with extremity weakness and impairment in mobility, is the most common presentation, occurring in about 70% of patients.2 Bulbar onset with oropharyngeal muscle involvement affecting swallowing and speech occurs in about 25% of cases.2 In addition to motor impairment, degeneration in the frontal and temporal lobes, resulting in cognitive or behavioural impairments, occurs in up to 50% of patients.3 Over time, strength progressively declines, and patients typically die from respiratory failure within 5 years of diagnosis.2 Despite increased research efforts in recent years, treatment options for ALS remain limited, and patient care is focused primarily on managing symptoms and optimizing function and quality of life.2
An estimated 3000 Canadians are currently living with ALS.4,5 Advocacy groups and clinicians caring for patients with ALS have strongly supported the development of best practice recommendations for the care and management of these patients in Canada. Although ALS clinical practice guidelines have been published in the United States6,7 and in Europe,8,9 to date there have been no published guidelines explicitly for the care of patients with ALS in Canada.
In addition to providing an update on the evolving standard of care in ALS, the best practice recommendations in this guideline serve to address several issues important to Canadians, such as caregiver support, medication alignment and medical assistance in dying (MAiD). Developing the first Canadian ALS guideline is a critical step in an iterative process whereby these recommendations can be updated as evidence evolves, and research priorities can be identified and prioritized to fill knowledge gaps.
Because the rigorous standards of evidence-based medical recommendations are not met in most areas of ALS care, many of the recommendations presented are expert consensus on good practice. Typically, symptom management in ALS is extrapolated from evidence in other disease states. The recommendations presented in this guideline are based on best available evidence and expert consensus on best practices, and thus reflect the real-life experiences of Canadian clinicians caring for patients with ALS. This article is a summary of the full guideline, which is available on the ALS Canada website (www.als.ca/bpr-appendix).
Scope
The purpose of this guideline is to provide ALS clinicians, allied health professionals and primary care providers with best practice recommendations for the care and management of patients living with ALS in Canada, inclusive of all genders, ages and stages of the disease. This guideline is intended to develop a national standard to improve quality of care for patients, families and caregivers living with ALS. Advocacy groups (e.g., ALS Society of Canada [ALS Canada], provincial ALS societies), health authorities, governments and policy-makers will be better able to establish benchmarks and advocate for standards of care.
Recommendations
The care and management of patients with ALS should always be patient focused, with attention to holistic and emotional aspects of well-being. It is the patient who ultimately decides on their treatment; this includes the option of declining interventions.
The recommendations for the management of patients with ALS in Canada are in Table 1, grouped by topic, and indicating the level of evidence. If evidence was insufficient or absent for a key question, we made recommendations based on expert consensus through review of the available literature and clinical experience in ALS or extrapolated from treatment of other more common diseases.
Recommendations with level of evidence grade for the management of patients with amyotrophic lateral sclerosis*
The following sections provide background and expand on selected recommendations with supporting evidence. The evidence tables in Appendix C of the full guideline (available at www.als.ca/bpr-appendix) provide details of supporting evidence for all recommendations listed in Table 1. For a complete discussion of the evidence supporting the recommendations, please see Appendix A (www.als.ca/bpr-appendix).
Communication of diagnosis
The manner in which the diagnosis of ALS is delivered is a source of discontent for many patients and caregivers.10,11 Recommendations have been formulated that outline a comprehensive approach to diagnosis delivery in the context of ALS.8 One of the most important concepts for clinicians to consider is tailoring the diagnosis delivery to the individual needs of the patient. If a patient is overwhelmed by the diagnosis of ALS, then the diagnosis could be delivered in a stepwise fashion, without divulging all of the information at once.12 Conversely, patients may feel that they did not receive enough information when receiving their diagnosis. Patients and caregivers wish to be informed about current research, treatments and prognosis when receiving a diagnosis of ALS.13
In a study of satisfaction with the manner of disclosure of the diagnosis of ALS, 41% of patients indicated that they received insufficient information, and one-third stated that they were not given a contact for follow-up.10 Furthermore, about 75% of patients and caregivers had questions that arose immediately after they received the initial diagnosis.11 These findings highlight the need for clinicians to address sources of information, community support and provide timely follow-up after the diagnosis is first discussed. Patients report better satisfaction with the delivery of an ALS diagnosis if they believe that the clinician has understood their feelings.13 An additional source of frustration for patients was the delay in receiving confirmation of a diagnosis, including wait times to see an ALS specialist.10 The working group agreed that a maximum wait time of 4 weeks for a consultation to confirm a diagnosis of ALS was reasonable.
Disease-modifying therapies
Health Canada approved riluzole as a treatment for ALS in 2000. Based on a class I meta-analysis of 4 randomized controlled trials (RCTs), riluzole has a modest benefit on survival compared with placebo, with a hazard ratio (HR) of 0.84 (95% confidence interval [CI] 0.698 to 0.997), representing a 9% gain in annual probability of survival. This translates to an increase in median survival from 11.8 to 14.8 months.14 Recent registry-based cohort studies (all class III) have estimated an improvement in median survival with riluzole treatment of 7.3 months,15 10 months16 or 12 months,17 but other studies have found no effect on survival.18–21 Findings from other class III cohort studies reported HR estimates of 0.34,22 0.71,23 0.7924 and 0.81,25 which translates to an estimated absolute increase in annual survival that ranges from 10% to 50%.
No controlled trials have examined whether riluzole extends life at a specific stage or all stages of ALS. A post-hoc analysis of the original dose-ranging study suggests that riluzole may be effective at prolonging survival only at later disease stages (defined by nutritional or respiratory failure sufficient to require intervention),26 but results from other cohort studies differ, showing that it may be effective only at earlier stages,27 or that its effect on survival is short lived.28 Nevertheless, decades of experience worldwide have shown riluzole to be generally well tolerated with prolonged use and with regular monitoring of liver enzymes and blood counts, as well as screening for nausea and fatigue (class I).14
Health Canada approved edaravone to treat ALS in October 2018. A single class I study in a generalized ALS population did not demonstrate overall benefit of edaravone in slowing progression of the ALS Functional Rating Scale–Revised (ALSFRS-R) score over 6 months29 but did suggest benefit in a subgroup of patients (see Table 1 for characteristics of this subgroup). This beneficial effect on the slowing of the progression of the ALSFRS-R score was subsequently confirmed in a second class I study that restricted recruitment to patients with characteristics of the subgroup from the first study.30 The second study demonstrated a mean reduction in the change in ALSFRS-R score over 6 months of 2.49 (95% CI 0.99 to 3.98). At this time, the available evidence suggests a level B evidence rating of “probably effective” in a select group of patients with ALS.
Multidisciplinary care
Patients with ALS should be regularly followed by a multidisciplinary ALS clinic, along with their primary care provider. Multidisciplinary care should be delivered through a team-based approach, with physicians and other health professionals addressing a broad range of issues, including communication, nutrition, swallowing, mobility, activities of daily living, respiratory care, cognition, psychosocial issues, medical management and end-of-life care. Patients followed through a multidisciplinary clinic have been shown to have better outcomes, including improved survival, fewer hospital admissions, increased use of adaptive equipment and enhanced quality of life, than those not followed in a multidisciplinary clinic.31–33 One prospective cohort study showed that patients followed in a multidisciplinary clinic lived 7.5 months longer than those followed in a general neurology clinic.32
Telemedicine and telehealth monitoring are feasible and may be able to supplement clinic-based multidisciplinary care.34 Management of patients with ALS should be a collaboration between the family physician and the ALS clinic, with the ALS clinic staff available for remote consultation between patient visits.
Respiratory management
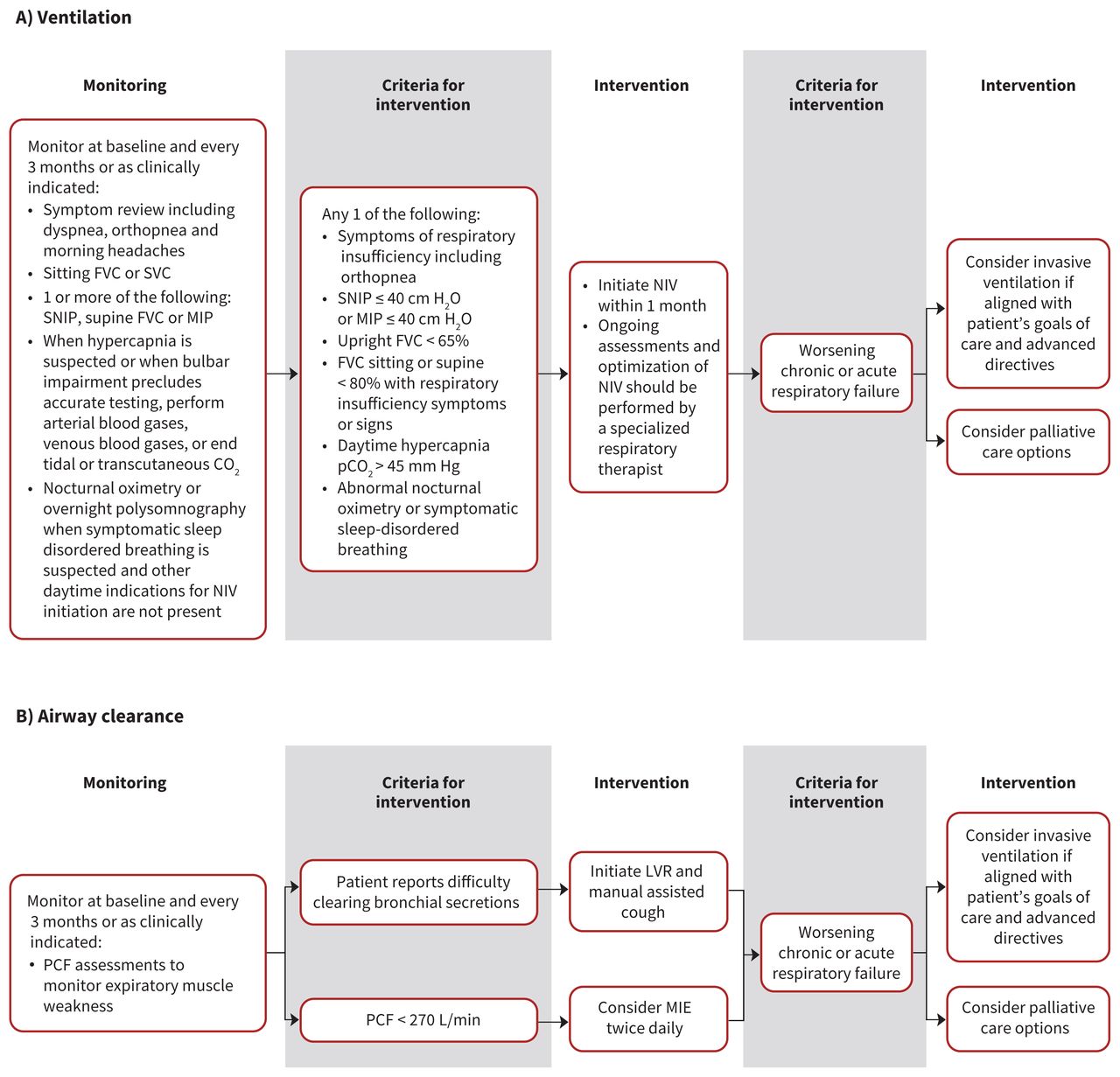
The Canadian Thoracic Society (CTS) guideline group recently reviewed the respiratory management of patients with ALS on home mechanical ventilation.35 We decided to make our recommendations on respiratory management (Table 1, Figure 1) consistent with the CTS guideline. We made a few additions to the CTS recommendations, including a statement on avoiding the use of oxygen for respiratory symptoms in patients with ALS, timing of initiation of interventions and managing secretions. We also thought it was important to adjust the minimum forced vital capacity (FVC) criterion for initiation of noninvasive ventilation in asymptomatic patients to 65% of predicted, from the CTS recommendation of 50%, because available evidence suggests early initiation improves survival.36 Patients with an FVC of greater than 65% predicted can be started on noninvasive ventilation if any of the other initiation criteria are met, as consistent with the CTS guideline. Our group also unanimously agreed that if criteria for initiation of noninvasive ventilation are met, patients should be initiated on noninvasive ventilation within 4 weeks.

Respiratory decision tree: Summary of recommendations for respiratory management in patients with amyotrophic lateral sclerosis (ALS), including ventilation (A) and airway clearance (B). Note: FVC = forced vital capacity, H2O = water, LVR = lung volume recruitment, MIE = mechanical insufflation–exsufflation, MIP = maximal inspiratory pressure, NIV = noninvasive ventilation, PCF = peak cough flow, pCO2 = partial pressure of carbon dioxide, SNIP = sniff nasal inspiratory pressure, SVC = slow vital capacity.
It is important to acknowledge that noninvasive ventilation can change the natural disease trajectory of ALS. For example, increasing reliance on noninvasive ventilation converts it into life-support technology. In patients reliant on noninvasive ventilation, natural death may not occur while using the technology; death may occur only if there is an active decision to discontinue the ventilation support. Patients should be counselled that they may need to take an active decision as to the timing of discontinuing the ventilatory support, unless they wish prolonged survival.
Difficulty with secretion management is common among persons living with ALS, and is a cause of distress, reduced quality of life and impairment of respiratory function. The CTS guideline did not explicitly address airway clearance management. To address this important issue, we reviewed available evidence and clinical experience. We recommend that lung volume recruitment techniques be introduced whenever patients present with symptoms of retained airway secretions or difficulty in clearing secretions. Such techniques can be combined with manual assisted coughing and be performed independently by patients or with assistance of care providers. If patients develop impaired peak cough flow (< 270 L/min), then mechanical insufflation–exsufflation twice daily should be considered for secretion clearance, and more frequently during an acute respiratory infection.
We also attained consensus that providing adequate in-home respiratory support of noninvasive ventilation and mechanical insufflation–exsufflation for education, titration and troubleshooting is essential, regardless of whether the patient resides in their own home, long-term care facility or hospice.
Nutrition management
The nutrition recommendations (Table 1, Figure 2) largely follow those outlined in the AAN guideline.6 Differences from the AAN recommendations include the addition of an expert consensus statement on the 4-week maximum allowable delay for a feeding tube insertion after criteria have been met, and a statement on the availability of appropriate follow-up after insertion for immediate or late complications. The recommendations also include a statement about nutritional components, and note that high-calorie diets can be used to improve nutritional indicators and possibly survival.37,38 High-calorie and high-carbohydrate diets may be better than high-calorie and high-fat diets.39

Nutrition decision tree: Summary of recommendations for nutritional management in patients with amyotrophic lateral sclerosis (ALS). Note: BMI = body mass index, FEES = fibre-optic endoscopic evaluation of swallowing, FVC = forced vital capacity, MBS = modified barium swallow, NG = nasogastric tube, PEG = percutaneous endoscopy gastrostomy, RIG = radiologically inserted gastrostomy, TDEE = total daily energy expenditure.
Venous thromboembolism
There is likely an increased risk of VTE in patients with ALS.40,41 The risk appears heightened in ALS with leg onset and in patients with poor mobility.40 Despite this elevated risk, there are no studies to support primary VTE prophylaxis. At this time, primary VTE prophylaxis is not recommended because the risk–benefit ratio of potential adverse consequences from falls versus VTE prevention in patients with ALS is uncertain.
Medication alignment
When patients come to an ALS clinic, they are often on multiple medications. Some of these medications may be considered nonessential, particularly considering the average survival of patients with ALS. Through expert consensus, we developed several statements that address the need for regular review of the medications that a patient is taking and suggest discontinuation of any nonessential medications that are not providing symptomatic relief or appropriate therapeutic benefit in the context of an individual patient’s expected survival.
Symptom management
Patients with ALS often have multiple uncomfortable symptoms that severely impair quality of life, including pain, fasciculations, sialorrhea, pseudobulbar affect, spasticity, cramps, depression, anxiety, insomnia and fatigue. Several clinical trials have explored treatment options for sialorrhea42 and pseudobulbar affect.43 However, management of most ALS symptoms has not been rigorously evaluated. As a consequence, most of the recommendations for symptom management were decided by expert consensus and supported by treatment suggestions made in the ALS and palliative care literature. Cost and access to treatments affected our ordering of the recommendations and were weighted more highly than direct evidence if an evidence-supported treatment was expensive. Our recommendations did not include the option of cannabis to treat specific ALS symptoms, because of lack of evidence in the literature. However, the working group is aware that cannabis is being used to manage several ALS symptoms.
Dysarthria
The ability to communicate thoughts and needs to others is vitally important to individuals. ALS often impairs the ability to communicate verbally because of dysarthria.2 Multiple available interventions can be initiated to support communication, including low-tech options, such as letter- or picture-boards, and high-tech options, such as speech synthesizers and eye-gaze tracking. As individuals with ALS experience loss of function, some modes of communication may no longer be viable. Providing access to different modes of communication, including social media, can allow independence, participation and better quality of life.44 Communication devices may also benefit caregivers, as the burden on caregivers was found to be reduced when patients used an eye-tracking assistive device.45
Exercise
Research on exercise in ALS has not demonstrated harm and some evidence has suggested that there is a potential benefit for patients in terms of function and quality of life.46,47 A personalized exercise program, including strength and aerobic training, should be encouraged for patients who are able to participate. A regular stretching and range-of-motion program is recommended for management of spasticity and pain, and prevention of contractures.
Cognition and behaviour
Detectable frontotemporal dysfunction can occur in about 50% of patients with ALS.3 The frontotemporal dysfunction can present with cognitive impairment or behavioural impairment, which in 20% of patients is severe enough to reach criteria for dementia.3 Although there are many tools available to screen for cognitive or behavioural impairment, there is no standard tool to use. At this time, there are no effective drug treatments for cognitive or behavioural impairment in ALS. A multidisciplinary approach can be considered to manage particularly problematic behaviours.
The presence of executive dysfunction or dementia in ALS is associated with poor survival.48 The presence of cognitive or behavioural impairment should not necessarily preclude implementing the recommendations for noninvasive ventilation or gastrostomy insertion. However, the challenges of intervention compliance with cognitive or behavioural impairment should be discussed with the patient and family before deciding to proceed with an intervention.
Caregivers
Informal caregivers are affected by caring for the person with ALS. Many studies have demonstrated the impact of ALS on caregiver quality of life and the correlates of caregiver burden,49 but also the value of caregiving.50 Advanced disability (a low ALSFRS-R score) and cognitive impairment increase caregiver strain.51 Interventions to mitigate the impact on caregivers have been insufficiently studied to make specific recommendations. People with ALS are aware of and affected by the burden on their caregivers.52 Health care providers, therefore, need to be attentive to the physical and emotional well-being of the caregivers, and involve them in planning for the impact of ALS on both the patient and themselves.
Palliative care
Expert opinion supports early integration of palliative care for patients with ALS.8,53 However, palliative and end-of-life care are sensitive topics and variably received by patients.54 Therefore, early introduction of palliative care must be initiated with consideration of the patient’s evolving needs and expectations.55 At the very least, experts have advocated that it is appropriate to initiate discussions about palliative care if the topic is raised by patients or caregivers, and if there are indications of advanced disease or disability.53
Advance care planning helps establish care preferences before the disease is advanced and communication is impaired. There is evidence to suggest that these discussions are best initiated when the patient has accepted that death will eventually occur.56 However, there is a general reluctance among clinicians to broach the topic, as it may be perceived to indicate the imminence of death.57 Standardized tools for advance care planning are thought to be useful for stimulating these discussions, rather than for generating specifics of an advanced directive.56,58,59 Thus, discussions may be integrated into routine ALS follow-up to invite open conversation, and should take into account the patient’s readiness and style of decision-making.
Medical assistance in dying (MAiD) was legalized in Canada in 2016. We have made specific recommendations as to how requests for MAiD should be addressed, both to support patient choice at end of life, and to provide guidance in this new practice, which may be a source of clinical uncertainty and discomfort to some practitioners.
We also present recommendations on the potential option of organ donation at the time of death and the process that should be followed for donation.
Methods
The concept for this guideline was concurrently fostered by ALS Canada and Canadian ALS clinicians within the Canadian ALS Research Network (CALS; now merged with ALS Canada). The guideline was developed using the Guideline International Network–McMaster Guideline Development Checklist,60 for guidance on all aspects of guideline development, including planning, formulation of recommendations, implementation and evaluation. A complete description of the guideline methodology is available in the full guideline (www.als.ca/bpr-appendix).
Guideline panel composition
A working group of 13 Canadian ALS clinicians (the authors), chaired by C.S., led the development of this guideline. Neurologists and physiatrists who were active in the Canadian ALS Research Network and could represent the geographic diversity of Canada were invited to participate in the working group. Clinicians with previous experience with guideline development were particularly encouraged to participate. The working group also included a gastroenterologist (D.L.) and a respirologist (A.T.) with ALS expertise. Early in the guideline development process, 2 other Canadian ALS clinicians were involved, but they removed themselves from the project because of the time commitments required.
Selection of key questions
In 2014, we selected clinical questions of interest for the guideline by surveying clinicians and staff at all 19 Canadian multidisciplinary ALS clinics via an emailed survey. The survey included a list of the key questions used to develop the American Academy of Neurology (AAN) Practice Parameters6,7 and European Network for the Cure of ALS guideline,8 as well as additional questions that members of the working group had derived based on their own clinical experience. We asked survey participants to rate the importance of these questions for inclusion in the guideline.
Questions included in the literature review were those questions rated highly by participants on the survey; the working group further refined these questions. The selected clinical questions were grouped by topic including communication of diagnosis, disease-modifying therapy, multidisciplinary care, respiratory management, nutrition management, symptom management, cognitive impairment, risk of venous thromboembolism (VTE), exercise, palliative care and caregiver support.
Literature search
In 2015, the Centre for Effective Practice, a consulting firm with substantial guideline development experience, conducted literature searches for the selected clinical questions using MEDLINE, Embase and CINAHL databases. The centre developed the search terms for each clinical question through review of the search terms that had been used for the AAN guideline6,7 and in consultation with the working group. The centre performed a second literature search in December 2018 to search for papers published after the initial search in 2015. For clinical questions addressed in the AAN guideline, literature searches were restricted to publications dated from 2007 to December 2018. For new clinical questions that had not been addressed in the AAN guideline or European Network for the Cure of ALS guideline, literature searches were restricted to publications dated from 1998 to December 2018. The search strategies are available in Appendix A of the full guideline (www.als.ca/bpr-appendix).
Quality assessment
The working group was divided into topic groups, with 2 members per group. For questions grouped under a major topic, 2 members of each topic group screened the retrieved abstracts separately based on the inclusion criteria and relevance to the clinical question. Inclusion criteria included published ALS guidelines, ventilation guidelines, RCTs, case–control studies, cohort studies, systematic reviews and meta-analyses. Publications had to be published in English or French and available in full text. Single- case reports, review articles, publications available only in abstract or proceeding forums, and thesis data not published elsewhere were excluded. Publications felt by at least 1 of the abstract reviewers to meet the inclusion criteria were reviewed in full by the topic group for inclusion criteria and data quality and assigned a class of evidence based on criteria modified from the AAN Clinical Practice Guideline Process Manual (2011 Edition) to rate therapeutic studies (Box 1). The evidence tables are available in Appendix B of the full guideline (www.als.ca/bpr-appendix).
| Class | Description |
|---|---|
| I |
|
| II |
|
| III |
|
| IV |
|
* Modified with permission from AAN (American Academy of Neurology). 2011. Clinical practice guideline process manual, 2011 Ed. St. Paul (MN): The American Academy of Neurology; 2011. Available online at www.aan.com/siteassets/home-page/policy-and-guidelines/guidelines/about-guidelines/11guidelinedevmanual_v408_web.pdf. Accessed 2020 Sept. 11.
Development of recommendations
The working group met regularly at face-to-face meetings at least annually in Toronto, and through regular group teleconferences to discuss the specifics of guideline statements. Each topic group drafted preliminary guideline statements for each clinical question after considering previously published guideline statements6–8 and updated evidence.
The working group reviewed these draft statements and refined them on an iterative basis, ideally until consensus was obtained. If consensus could not be reached among the working group, we agreed that a decision would be made based on a two-thirds majority (66%) vote. However, there was consensus on all statements and so no vote was held.
We assigned each statement a level of evidence, which included the option of expert consensus (Box 2). The working group felt strongly that in the absence of published evidence, best practice recommendations based on expert consensus should be included, rather than no recommendation provided. Given that there is limited evidence from clinical trials to direct care in ALS, the working group members thought it important that the recommendations be a practical guide to the care of patients with ALS, rather than simply a review of the evidence. We made expert consensus statements based on nonclinical trial literature in ALS, evidence in other diseases or current Canadian ALS clinical practice. We discussed the order of the statements in the recommendations table at length to reflect their clinical importance and the order in which a practitioner would consider interventions when caring for patients.
| Level | Type of evidence |
|---|---|
| A | At least 2 consistent class I studies |
| B | At least 1 class I study or 2 consistent class II studies |
| C | At least 1 class II study or 2 consistent class III studies |
| Expert consensus | Consensus among Canadian amyotrophic lateral sclerosis clinical experts where evidence meeting criteria for Level A through Level C is lacking |
* See Box 1 for definitions of study classes.
After the statements we developed for the respiratory questions had obtained consensus support from our working group, the Canadian Thoracic Society (CTS) published a guideline on the respiratory care of patients with ALS.35 The working group decided it was important for our recommendations to be consistent with this guideline. To accomplish this, we compared each of the recommendations in the CTS guideline with our draft statements. Our working group accepted most of the CTS recommendations without changes, apart from slight wording alterations for consistency. We asked the CTS ALS committee to review our suggested statements, including those expert consensus statements where questions deemed important in our survey had not been addressed by the CTS guideline; their feedback led to some minor changes in our wording.
Review process
We developed an executive summary of the guideline statements and the working group reviewed it. When the working group was satisfied with the recommendation statements, including the wording, order and evidence ranking, we emailed this executive draft summary to members of the Canadian ALS Research Network (which includes all multidisciplinary ALS clinics in Canada) and topic experts external to the working group (i.e., with expertise in gastroenterology, respirology, palliative care and physiatry) for open-ended feedback. We asked the ALS clinics to share the executive draft summary with their allied health staff and request additional open-ended feedback from them as well. The working group discussed each comment received to determine whether changes were required to the recommendation statements and if so, how the statements should be revised.
Using the revised executive draft summary, a second round of external review followed, in which we asked key stakeholders within each provincial ALS society to participate. We emailed the revised executive draft summary to each of the provincial ALS societies along with an attached survey with open-ended questions. We asked each society to solicit feedback from its members, including 1 patient living with ALS in its province. All comments received were individually considered by the working group and changes were implemented at its discretion through a robust discussion about the feedback. The changes made according to the feedback received involved wording changes for the most part. We made no substantial changes.
We prepared a complete version of the guideline and all working group members reviewed it for final approval.
Management of competing interests
All members of the working group performed their tasks voluntarily and did not receive honoraria for their involvement. ALS Canada and the Canadian ALS Research Network funded the development of the guideline, including travel for face-to-face meetings and preparation of the manuscript for publication.
ALS Canada is a grassroots donor–funded organization and part of the funding for this project came from donations during the Ice Bucket Challenge. ALS Canada assisted with logistic support but did not contribute to the content of the recommendations. The Canadian ALS Research Network was a nonprofit organization of ALS clinicians and researchers formed to increase clinical ALS research in Canada and funded by stipends given by biotechnology companies to review clinical trial proposals for Canadian ALS clinics (it has subsequently merged with ALS Canada). Although members of CALS participated in the development of the guideline, CALS had no role in approving guideline recommendations.
We discussed competing interest management during the planning phase of the guideline; competing interests were defined as a financial relationship with a company. At that time, there was only 1 drug approved by Health Canada for the treatment of ALS: riluzole. None of the working group members had conflicts related to the drug riluzole, which has been available for more than 20 years. We solicited other potential conflicts of interest from the working group at the beginning of this project, and no conflicts were present.
In 2017, during the guideline development process, the US Food and Drug Administration approved edaravone to treat ALS. Its manufacturer, Mitsubishi Tanabe Pharma, sponsored scientific advisory committees regarding the use of edaravone in patients with ALS in Canada. Health Canada approved the drug in October 2018 and it became commercially available in Canada in November 2019. Some working group members sat on the Mitsubishi Tanabe Pharma scientific advisory committees for edaravone (C.S., M.C., A.I., W.J., C.O., K.S., L Z.), which they disclosed to the working group. All members of the working group discussed at length the statements in this guideline regarding edaravone. During review of the draft guideline, feedback from members of the Canadian ALS Research Network and key stakeholders regarding the edaravone statements was deliberated by working group members who did not have conflicts of interest with Mitsubishi Tanabe Pharma, defined as having received any honoraria from the company. Final decisions regarding the edaravone statements were made by working group members without potential conflicts. Other than edaravone, there are no other potential conflicts with the statements in this guideline.
Implementation
These best practice recommendations are a resource to guide the care of patients with ALS across Canada. The guideline will be made publicly accessible through the ALS Canada website (www.als.ca). ALS Canada will also support the dissemination of the guideline among members of the ALS community, including clinicians, allied health professionals, researchers, patients and their caregivers, through distribution to provincial ALS societies, the Canadian ALS Research Network and attendees of the annual ALS Canada Research Forum. Directors of ALS clinics and ALS clinicians will be encouraged to present the guideline to their clinic teams and relevant stakeholders within their communities. ALS Canada will assist the guideline authors with producing 1-page summary documents of some key clinical areas of the guideline for dissemination to stakeholders.
The working group would support a health impact project assessing patient survival, patient-perceived quality of life and other specific outcomes after the implementation of the guideline compared with before its publication.
The working group expects that evidence to support ALS management will evolve over time and anticipates that the recommendations will have to be revised approximately every 5 years.
Other guidelines
Several ALS clinical practice guidelines have been published in countries other than Canada, including the AAN Practice Parameters (2009),6,7 the European Federation of Neurological Societies guideline on the clinical management of amyotrophic lateral sclerosis (2012),8 and the motor neurone disease assessment and management guideline developed by England’s National Institute for Health and Care Excellence (2016).9
One of the goals for the Canadian guideline was to update the existing North American guidelines, specifically the 2009 AAN recommendations.6,7 As described in the Methods section, literature searches for this Canadian guideline on clinical questions addressed in the 2009 AAN recommendations were restricted to new evidence only (i.e., after 2007), and all evidence was classified using AAN criteria.
In the AAN guideline, recommendations had to be supported by evidence; thus, no guidance was provided in the absence of evidence (e.g., using expert consensus). In contrast, the European Federation of Neurological Societies guideline provided consensus recommendations in the absence of evidence. We also resolved to offer guidance based on expert consensus in the absence of evidence.
Another goal for the Canadian guideline was to address ALS issues not covered in other guidelines. The European Federation of Neurological Societies guideline did not address several issues for patients with ALS that are important in Canada, such as medication alignment and MAiD. Similarly, guidance on some ALS issues, such as disease-modifying treatments and exercise, was not provided in the National Institute for Health and Care Excellence guideline.
As discussed earlier, the CTS published a guideline on home mechanical ventilation for patients with ALS in early 2019.35 In collaboration with the CTS, we ensured that our recommendations for respiratory management were consistent with recommendations in the CTS guideline, but added some consensus recommendations (e.g., on airway clearance).
Gaps in knowledge
This guideline confirms that high-quality evidence is lacking for most topics in ALS management; most recommendations provided are based on expert consensus among the working group. The need for further research in ALS management remains, and more evidence-based recommendations will be critical for improving the standards of patient care in Canada and internationally. This guideline can help point the clinical research community, nationally and internationally, to areas of research priorities on disease management.
We acknowledge that we were not able to cover all topics of ALS management in this guideline and that subsequent revisions could include topics not currently covered.
Conclusion
We hope that the development of the first Canadian ALS guideline is an important step forward for improving the lives of patients with ALS living in Canada. The predominance of expert consensus statements relative to evidence-based statements in this guideline not only highlights the need for more research in ALS management but also emphasizes the challenges ALS clinicians face in managing patients with a severe disabling disease. This guideline will enable ALS clinics across Canada to meet a common national standard, and to adapt as this standard continues to evolve over time. In doing so, ALS clinicians can offer the best possible care to their patients and help them to navigate this exceedingly complex and devastating disease.
Acknowledgements
The writing group thanks ALS Canada for its ongoing support for the development of these guidelines. The authors also acknowledge Ms. Vanessa Blount, who helped with the initial coordination of the guidelines, Dr. David Taylor and Ms. Colleen Doyle for logistical support, and Ms. Trisha Rao, who helped with the editing of the document. The Canadian ALS Research Network and Federation partners also provided critical feedback with respect to the wording of the guidelines and manuscript. Dr. Ikhlass Salem Haj, Dr. Lawrence Korngut and Dr. Hannah Briemberg also provided substantial assistance in the development of these recommendations.
Footnotes
This guideline is available in French at www.cmaj.ca/lookup/doi/10.1503/cmaj.191721-f
CMAJ Podcasts: author interview (in English) at www.cmaj.ca/lookup/doi/10.1503/cmaj.191721/tab-related-content
Competing interests: Christen Shoesmith, Aaron Izenberg, Wendy Johnston, Colleen O’Connell, Kerri Schellenberg and Lorne Zinman were members of a scientific advisory committee for Radicava (edaravone; Mitsubishi Tanabe Pharma Canada). Christen Shoesmith reports being a site principal investigator for several multicentre amyotrophic lateral sclerosis (ALS) clinical trials. In the last 36 months, Dr. Shoesmith has participated in clinical trials sponsored by Biogen, Cytokinetics, ALS Pharma and Orphazyme. Marvin Chum reports receiving a grant from Bernice Ramsay ALS Clinical Research Fellowship, outside the submitted work. Aaron Izenberg reports receiving personal fees from Biogen, Roche, Alnylam, Genzyme, Takeda and Mitsubishi Tanabe Pharma, outside the submitted work. No other competing interests were declared.
This article has been peer reviewed.
Contributors: All of the authors contributed to the conception and design of the work, and the acquisition, analysis and interpretation of data. All of the authors drafted the manuscript, revised it critically for important intellectual content, gave final approval of the version to be published and agreed to be accountable for all aspects of the work.
Funding: Funding for the development of the recommendations was provided by the ALS Society of Canada and the Canadian ALS Research Network.
References
In this issue

Article extras
Article tools



{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- The complexity of multidisciplinary respiratory care in amyotrophic lateral sclerosis
- Amyotrophic Lateral Sclerosis Quality Measurement Set 2022 Update: Quality Improvement in Neurology
- Increased Risk of Venous Thromboembolism in Patients With Amyotrophic Lateral Sclerosis: Results From a US Insurance Claims Database Study
More in this TOC Section
Similar Articles
Collections


Podcast